PRIMA FACOLTA' DI MEDICINA E CHIRURGIA - CLM "B"
SAPIENZA UNIVERSITA' DI ROMA
Si definisce soluzione una miscela di sostanze che condividono un'unica fase termodinamica. Le soluzioni possono trovarsi allo stato gassoso (e allora sono piu' spesso definite miscele gassose); per la medicina la miscela gassosa piu' importante e' l'aria. Possono trovarsi allo stato liquido e' questo e' il caso piu' frequente e il piu' importante per la medicina. Possono infine trovarsi allo stato solido, come avviene per le leghe metalliche, che hanno alcune applicazioni mediche (protesi, otturazioni dentarie, etc.).
Le soluzioni solide (essenzialmente le leghe metalliche e i cristalli misti dei sali isomorfogeni) hanno troppo poco interesse medico per giustificare una descrizione dettagliata; saranno quindi studiate soltanto le soluzioni liquide e le miscele gassose.
I COMPONENTI DELLE SOLUZIONI. Le soluzioni sono costituite da almeno due sostanze diverse, delle quali quella presente in maggiore quantita' prende il nome di solvente, l'altra (o le altre) quello di soluto (o soluti).
In genere lo stato di aggregazione della soluzione e' quello che il solvente avrebbe se fosse da solo nelle condizioni di temperatura e pressione considerate, ma esistono piccole variazioni: ad es. la soluzione liquida ha punti di congelamento e di ebollizione diversi da quelli del solvente puro e questo implica che in prossimita' del cambiamento di stato la soluzione ed il solvente puro possono differire per lo stato di aggregazione.
Si definisce concentrazione della soluzione (o del soluto) il rapporto tra la quantita' del soluto e la quantita' del solvente o della soluzione. Poiche' esistono varie unita' di misura per la quantita' di materia, esistono anche varie unita' di misura per la concentrazione:
1) percentuale peso/peso: grammi di soluto contenuti in 100 g di soluzione.
2) percentuale peso/volume: grammi di soluto contenuti in 100 ml di soluzione.
3) percentuale volume/volume: ml di soluto contenuti in 100 ml di soluzione (applicata ai soluti che nel loro stato puro sono liquidi o gassosi).
4) molarita': moli di soluto contenuti in 1 litro di soluzione.
5) normalita': numero di equivalenti di soluto contenuti in 1 litro di soluzione.
6) molalita': moli di soluto contenuti in 1 kg di solvente.
7) frazione molare: rapporto fra le moli di soluto e la somma del numero di moli di ciascun componente della soluzione (solvente piu' ciascuno dei soluti).
Per la medicina la miscela gassosa piu' importante e' l'aria, una miscela di azoto, ossigeno ed altri gas. L'aria contiene una quantita' variabile di vapore acqueo (in frazione molare < 0,05); eliminato questo, le frazioni molari dei componenti dell'aria secca sono: N2 = 0,79; O2 = 0,20; altri gas = 0,01.
PRESSIONE PARZIALE. In una miscela di gas perfetti, ogni molecola si comporta in modo indipendente da ogni altra e ciascun gas contribuisce alla pressione totale della miscela in ragione del suo numero di moli.
Di conseguenza e' possibile calcolare la pressione parziale di ciascun gas, cioe' la pressione che quel componente eserciterebbe se fosse solo ad occupare il volume a disposizione della miscela:
Naturalmente la somma delle pressioni parziali e' pari alla pressione totale della miscela:
Si puo' facilmente dimostrare che il rapporto tra la pressione parziale e la pressione totale e' uguale alla frazione molare:
VOLUME PARZIALE. In analogia con la pressione parziale si definisce il volume parziale come il volume che ciascun componente di una miscela gassosa occuperebbe se fosse solo ad una pressione pari alla pressione totale della miscela:
Naturalmente la somma dei volumi parziali e' pari al volume totale della miscela:
Il volume parziale ha questo significato: la miscela potrebbe essere prodotta mescolando volumi di gas pari ai volumi parziali desiderati (tutti misurati alle stesse condizioni di temperatura e pressione); inoltre il rapporto tra il volume parziale ed il volume totale e' pari alla frazione molare:
USO DEI CONCETTI DI PRESSIONE E VOLUME PARZIALE. Si consideri il seguente esperimentoche ha lo scopo di determinare la frazione molare dell'ossigeno in una miscela gassosa. Si ha a disposizione un recipiente contenente la miscela e si puo' operare sia a volume che a pressione costante; in ogni caso devono essere misurati i due parametri (pressione e volume). Si introduce nel recipiente della limatura di zinco (senza permettere la fuoriuscita del gas!) in modo da consumare l'ossigeno (ma non gli altri gas) con la reazione 2 Zn + O2 --> 2 ZnO. Se l'esperimento e' condotto a volume costante si osservera' una diminuzione di pressione pari alla pressione parziale dell'ossigeno; se invece l'esperimento e' condotto a pressione costante si osservera' una diminuzione di volume pari alvolume parziale dell'ossigeno.
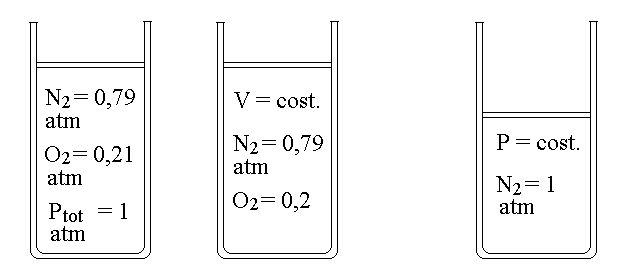
Figura 1: consumando l'ossigeno contenuto una miscela gassosa (quale l'aria) si possono osservare le pressioni o i volumi parziali dei gas rimanenti (e calcolare per differenza quelli dell'ossigeno consumato).
Per molte coppie soluto-solvente esiste una concentrazione massima, raggiunta la quale il soluto non e' piu' solubile e precipita formando il cosiddetto "corpo di fondo".
Ad esempio il comune sale da cucina (NaCl) e' solubile fino alla concentrazione di circa 6 M; pertanto se si prova a discioglerne una quantita' piu' grande di 34 g/ 100 ml si ottiene un corpo di fondo (100 ml e' circa mezzo bicchiere di acqua, provare per credere).
La massima concentrazione ottenibile di un soluto e' definita la sua solubilita' in quel solvente e la soluzione che la contiene e' definita satura. La solubilita' dipende dal composto considerato, dal solvente e dalla temperatura.
La relazione che correla la quantita' di soluto ed il volume della soluzione alla concentrazione:
C=n/V (7)
Questa relazione vale cosi' com'e' per la molarita' e puo' richiedere piccole variazioni per le altre unita' di misura della concentrazione (ad es. C=n/Kgsolvente per la molalita').
Oltre a permetterci di calcolare la concentrazione della soluzione puo' essere usata per calcolare il numero di moli di soluto contenute in un certo volume di una soluzione a concentrazione nota:
n=CxV (8)
Inoltre nella medicina questa relazione viene utilizzata per le determinazioni dei volumi:
V=n/C (9)
VOLUME MOLARE. Come per i gas perfetti anche per le soluzioni ci si puo' chiedere quale sia il volume molare del soluto, ovvero quale volume della soluzione contenga esattamente una mole di soluto. Il volume molare del soluto e' facilmente calcolabile dalla eq.9, uguagliando n ad 1: si ottiene che Vmolare = 1/C (docve C deve essere espressa in molarita').
DILUIZIONI. Una diluizione viene effettuata aggiungendo solvente puro ad una soluzione. Poiche' il numero di moli del soluto non cambia (in questo esperimento non viene ne' aggiunto ne' sottratto soluto ma soltanto solvente), la relazione fondamentale ci dice che:
n = C1 V1= C2 V2 (10)
Pertanto la concentrazione finale della soluzione risulta:
C2 = C1 V1 / V2 (11)
Il rapporto tra i volumi finale ed iniziale si chiama fattore di diluizione e poiche' V2 > V1, e' sempre maggiore dell'unita':
F = fattore di diluizione = V2 / V1 (12)
Vale quindi:
C2 = C1 / F (13)
MESCOLAMENTO. Quando si mescolano due soluzioni dello stesso soluto a diversa concentrazione se ne ottiene una terza a concentrazione intermedia; infatti:
n = C1 V1 + C2 V2 = C3 V3 (14)
V3 = V1 + V2 (15)
C3 = (C1 V1 + C2 V2) / (V1 + V2) (16)
DILUIZIONI SERIALI. Quando sia importante ottenere diluizioni molto spinte (ad es. negli esami sull'attivita' anticorpale del sangue) non e' pratico diluire il campione fino alla concentrazione desiderata in un unico passaggio, e si ricorre al metodo delle diluizioni seriali.
Si supponga di dover diluire 10.000 volte 1 ml di siero di sangue. Il fattore di diluizione richiesto corrisponde a V2=10.000 x 1 ml = 10 l. Chiaramente effettuare questa diluizione non e' pratico. Si procede quindi in questo modo: si diluisce il campione a 100 ml realizzando una prima diluizione con fattore F1=100; poi si preleva 1ml della soluzione ottenuta e lo si diluisce di nuovo a 100 ml, quindi ancora con fattore F2=100.
Se si fosse proceduto con unica diluizione e F=10.000 si sarebbe ottenuto:
Vin = 1 ml
Vfin = 10.000 ml = 10 l
Cfin = Cin / 10.000
Procedendo con il metodo descritto sopra e' stata invece realizzata una diluizione intermedia e si e' ottenuto:
Vin = 1 ml
Vint = 100 ml
Cint = Cin / 100
Vfin = 100 ml
Cfin = Cint / 100 = Cin / (100x100) = Cin / 10.000
Come si vede, il metodo delle diluizioni seriali produce lo stesso risultato della diluizione diretta ma e' piu' comodo e meno costoso.
Per generalizzare la formula, quando si realizza una serie di N diluizioni con fattore di diluizione costante F, la relazione tra la concentrazione finale e quella iniziale e' la seguente:
Il metodo delle diluizioni seriali e' usato in medicina per determinare il titolo (concentrazione) di alcune sostanze presenti nel sangue a basse concentrazioni quali ad esempio gli anticorpi specifici per alcune malattie. Il principio della determinazione e' semplice: si diluisce serialmente il siero di sangue del paziente e si determina qual e' la massima diluizione alla quale e' ancora possibile osservare la reazione caratteristica della sostanza cercata. Ad esempio un siero potrebbe avere un Titolo Anti Streptolisinico (TAS) pari a 10.000: questo vuol dire che l'anticorpo anti streptolisina e' ancora attivo dopo una diluizione con fattore 10.000 (realizzata in genere mediante una serie di diluizioni con fattore 5 o 10). Il valore TAS=10.000 e' considerato positivo, cioe' indica che il paziente presenta una infezione streptococcica recente o in corso, alla quale sta rispondendo con la sintesi degli anticorpi. Per contro un TAS=100 e' considerato negativo, cioe' indice di bassa concentrazione degli anticorpi e quindi di infezione assente o remota.
OMEOPATIA. La medicina omeopatica fu codificata da S. Hahnemann tra la fine del 1700 e l'inizio del 1800.
I principi sui quali si basa sono i seguenti:
1) il farmaco efficace e' quello che causa nel sano gli stessi sintomi che la malattia causa nel malato (legge dei simili; similia similibus curentur)
2) il farmaco deve essere somministrato in forma pura e deve essre adatto al paziente (individualizzazione): deve cioe' causare nel sano tutti (o la maggior parte) dei suoi sintomi e possibilmente soltanto quelli.
3) il farmaco aumenta di potenza se viene diluito serialmente (potentizzazione; legge delle diluizioni o degli infinitesimi). E' importante che ad ogni diluizione segua una energica agitazione (succussione).
Il principio n.3, che si interessa in questa sede, era applicato da Hahnemann con il metodo delle diluizioni seriali centesimali (indicate come C o CH). Hahnemann utilizzava le potenze "basse" (3-6C), "medie" (10-15C) e "alte" (fino a 30C ed oltre) ed e' facile calcolare (con la formula 17) che anche alle medie diluizioni viene superato il numero di Avogadro; di conseguenza la dose somministrata al paziente della gran parte dei farmaci omeopatici non contiene neppure una molecola della sostanza da cui prende il nome.
Una analisi critica piu' dettagliata dei principi dell'omeopatia e' disponibile su questo sito web.
Il nostro organismo e' composto da acqua per circa il 70% in peso; della parte non acquosa hanno massa significativa la matrice minerale dello scheletro (di fosfato di calcio) e il tessuto adiposo.
L'acqua del nostro organismo e' distribuita in tre compartimenti comunicanti: il liquido intracellulare; il liquido extracellulare ed il sangue.
In tutti e tre i compartimenti l'acqua e' il solvente di soluzioni complesse, di molti soluti micro- e macro-molecolari, tra i quali hanno particolare importanza:
- elettroliti (ioni sodio, potassio, calcio, magnesio, cloruro, bicarbonato ed altri)
- proteine
- glucosio
La concentrazione dei soluti nei liquidi biologici e' in genere mantenuta costante da complessi meccanismi di controllo, specifici per ogni soluto ed esercitati a livello renale, epatico, respiratorio, ormonale, etc.
Ad esempio, le concentrazioni medie dei principali soluti del plasma di sangue (per l'uomo sano) sono:
| ione sodio | 140 mM |
| ione cloruro | 100 mM |
| ione bicarbonato | 26 mM |
| ione potassio | 5 mM |
| ione calcio | 2,5 mM |
| glucosio | 80 mg / 100 ml |
| proteine | 7 g / 100ml |
Variazioni significative da questi valori sono spesso indice di malattie
IL SANGUE DELL'UOMO. Gli argomenti trattati in questa sezione del corso richiedono una minima familiarita' con la composizione dei liquidi biologici, in particolare del sangue. Il sangue intero come ottenuto da un prelievo arterioso o venoso e' un liquido denso e opaco, di intenso colore rosso. Raccolto con un anticoagulante e lasciato a se' si separa in due parti: una piu' pesante, sul fondo della provetta che lo contiene di colore rosso molto scuro: i globuli rossi; l'altra piu' leggera trasparente e di colore giallastro: il plasma. Sulla superficie di contatto tra le due componenti si nota a volte uno straterello bianco molto sottile (buffy coat) costituito da globuli bianchi e piastrine. Il volume dei globuli rossi corrisponde a circa il 45% del totale, quello del plasma al rimanente 55%. Il plasma e' una soluzione acquosa di proteine, sali ed altre sostanze; se gli si consente di coagulare se ne elimina una specifica proteina (la fibrina) e il liquido restante prende il nome di siero.
MISURE DEI VOLUMI DEI FLUIDI CORPOREI. In alcune circostanze il medico puo' desiderare di misurare non solo la concentrazione dei soluti ematici, ma anche il volume del sangue o degli altri compartimenti liquidi dell'organismo.
Questo tipo di misura viene effettuato sfruttando l'equazione 9.
Si procede come segue: si inietta per via endovenosa una sostanza non tossica e facile da identificare, definita tracciante (per il sangue si usa in genere albumina umana marcata con un isotopo radioattivo), in quantita' nota; poi, atteso un tempo sufficiente perche' questa si distribuisca in modo uniforme, si esegue un prelievo di sangue e si dosa la concentrazione della sostanza. A questo punto, noti n e C si determina V=n/C.
Il procedimento descritto puo' essere applicato per misurare il volume del sangue (la volemia), quello di tutto il liquido extracellulare (incluso quindi il sangue), e quello di tutti i liquidi presenti nell'organismo (total body water, TBW), variando semplicemente il tracciante usato (che deve ripartirsi in modo omogeneo in tutto il compartimento del quale si vuole misurare il volume) ed il tempo di attesa. Possibili sorgenti di errore: tracciante che non si ripartisce in modo omogeneo; tempi di attesa troppo brevi (che non consentono la distribuzione del tracciante in tutto il compartimento); tempi di attesa troppo lunghi (il tracciante viene eliminato, in genere per via renale, quindi si ha variazione di n).
MISURE DI FLUSSO. La determinazione di un flusso (volume/minuto) e' piu' complessa di quella del semplice volume. Un esempio tipico e' quello della misura del flusso ematico polmonare, rilevante per molte diagnosi. Per illustrare il procedimento immaginiamo una determinazione effettuata come segue.
Un paziente respira in uno spirometro, strumento che consente di determinare i volumi dell'aria inspirata ed espirata, ed il relativo contenuto di ossigeno. Un valore tipico per questa determinazione potrebbe essere:
aria inspirata 5 l/min, 1 atm, 25 gradi C, O2 20% in volume
nO2 inspirato/min = 5 l x 0,2 x 1 atm / (0,082 x 298 K) = 0,041 moli
aria espirata 5 l/min, 1 atm, 25 gradi C, O2 15% in volume
nO2 espirato/min = 5 l x 0,15 x 1 atm / (0,082 x 298 K) = 0,031 moli
Ossigeno trattenuto dall'organismo in un minuto = 0,041 - 0,031 = 0,01 moli
Nel corso della determinazioni dei volumi dell'aria inspirata ed espirata, il paziente viene sottoposto ad un prelievo di sangue arterioso e ad un prelievo di sangue venoso. Il sangue venoso e' quello che non ha ancora attraversato il polmone (quindi e' povero di ossigeno), quello arterioso e' quello che ha appena traversato il polmone (quindi e' ricco di ossigeno). In entrambi i campioni di sangue si determina il contenuto di ossigeno; questa determinazione non e' priva di specifiche difficolta', che pero' in questa sede vengono tarscurate. I valori medi tipici per le persone sane sono:
contenuto di O2 nel sangue arterioso = 0,01 moli/l
contenuto di O2 nel sangue venoso = 0,008 moli/l
differenza artero-venosa di O2 = 0,01-0,008 = 0,002 moli/l
La domanda che ci si pone e' la seguente: quanti litri di sangue devono attraversare i polmoni in un minuto per estrarre 0,01 moli di O2 (calcolate dalle misure effettuate sull'aria inspirata ed espirata) visto che ogni litro di sangue attraversando i polmoni si arricchisce di 0,002 moli del gas? La risposta e':
flusso polmonare = 0,01 moli/min / 0,002 moli/l = 5 l/min.
MISURE DI CLEARANCE. La clearance e' un flusso "teorico", immaginario, ma che fornisce una indicazione clinica rilevante sulla funzionalita' di alcuni organi (in genere del rene; occasionalmente del fegato o del polmone). Descriviamo qui la misura della clearance renale della creatinina (una misura comunemente utilizzata nella medicina) a titolo di esempio.
Mediante un prelievo venoso viene determinata la concentrazione della creatinina, un tracciante naturalmente presente nel sangue; il valore normale e' <1,2 mg/100 ml. Vengono raccolte le urine del paziente per un tempo sufficientemente lungo (mediante cateterismo vescicale e' possibile raccogliere l'urina prodotta in tempi brevi, ad es. in 15 min.; se si vuole evitare questa pratica e' opportuno raccogliere le urine su un periodo piu' lungo, ad es. 12 ore). Viene determinata la concentrazione della creatinina nelle urine. La clearance renale della sostanza puo' ora essere determinata come:
clearancecreatinina = Vurine Curine / Cplasmatica
Questa e' equazione costituisce una applicazione dell'eq. 10 e calcola in quale volume di plasma sanguigno era contenuta la creatinina escreta. Il medico avra' avuto cura naturalmente di riportare Vurine ad un volume/min. o ad un volume/h dividendo il volume totale dell'urina raccolta per il numero di minuti o di ore della raccolta, perche' la clearance ha le unita' di misura volume/tempo.
La clearance cosi' determinata non e' pero' un vero volume o flusso (al contrario del flusso polmonare calcolato sopra) perche' il valore calcolato risente del fatto che non tutto il sangue che attraversa il rene viene filtrato e depurato, e non tutto il soluto presente nel filtrato finisce poi nell'urina (alcuni soluti vengono riassorbiti, altri escreti dai tubuli renali). I valori medi per le persone sane sono i seguenti: flusso ematico renale circa 1,2 l/min (questo corrisponde ad un flusso di plasma di circa 650 ml/min); flusso del filtrato glomerulare circa 125 ml/min; riassorbimento dell'acqua oltre 99% e urina prodotta circa 1 ml/min. La creatinina non viene ne' assorbita ne' escreta dai tubili renali e quindi la sua clearance fisiologica media e' prossima al volume del filtrato (125 ml/min di plasma o 240 ml/min di sangue).
Ce ne sono di facili e di difficili. Un esempio di conversione facile e' quella da percentuale peso/volume a molarita' perche' le due unita' sono tra loro omogenee (infatti entrambe sono definite come quantita' di soluto diviso per volume di soluzione). Per effettuare questa conversione si procede come segue:
1) si moltiplica per 10 la percentuale peso/volume in modo da ottenere una misura di congentrazione in g/l (o mg/l; se si ottiene questa unita' di misura la si deve dividere per 1000 per trasformarla in g/l)
2) si divide per il peso molecolare per ottenere la molarita'.
Ad esempio la glicemia media dell'uomo sano e' di circa 90 mg% (o 90 mg/100 ml) ed il PM del glucosio e' 180 g/mole; pertanto la molarita' del glucosio nel sangue risulta:
90 mg% = 900 mg/l = 0,9 g/l
0,9 g/l /180 g/mole = 0,005 M = 5 mM
Un esempio di conversione difficile e' da molalita' o da percentuale peso/peso a molarita' (o viceversa), perche' queste unita' non sono omogenee tra loro. Per queste conversioni e' necessario conoscere la densita' della soluzione (d=peso/V).
Si consideri il seguente esempio: calcolare la molarita' della soluzione 3 m di ammoniaca (PM = 17 g/mole) che ha d= 0,95 g/ml.
Poiche' la concentrazione e' una grandezza intensiva, il ragionamento puo' essere condotto su qualunque quantita' di soluzione; per semplicita' considereremo quella quantita' di soluzione che contiene 1 kg di solvente perche' corrisponde alla definizione di molalita'. Il peso totale della quantita' di soluzione che contiene un 1 kg di solvente e' dato dalla somma dei pesi del solvente e del soluto (che ammonta a 3 moli /kg di solvente):
Pesosoluzione = 1000gH2O + 3 x 17 gNH3 = 1051 gsoluzione
Il volume si calcola a partire dal peso e dalla densita' con la formula V = peso/d; una volta trovato il volume la molarita' e' n/V:
V = 1051 g / 0,95 g/ml = 1106 ml = 1,106 l
C = 3 moli / 1,106 l = 2,71 M
Come e' costituita la soluzione al livello delle singole molecole del solvente e del soluto? Distinguiamo vari casi.
SOLUZIONI DI IONI. Gli elettroliti si disciolgono in solventi polari (ad es. l'acqua) dissociandosi negli ioni costituenti, ciascuno dei quali forma poi interazioni dipolo-dipolo con le molecole del solvente.
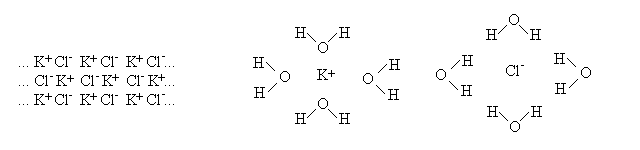
Figura 2: Legami ione-dipolo tra il cloruro di potassio e l'acqua.
Lo ione in soluzione e' circondato da una sfera di molecole di solvente (sfera di solvatazione; se il solvente e' l'acqua, sfera di idratazione) ordinate rispetto alla polarita'; queste ordinano a loro volta una seconda sefra di solvatazione piu' debolmente legata, ed eventualmente una terza.
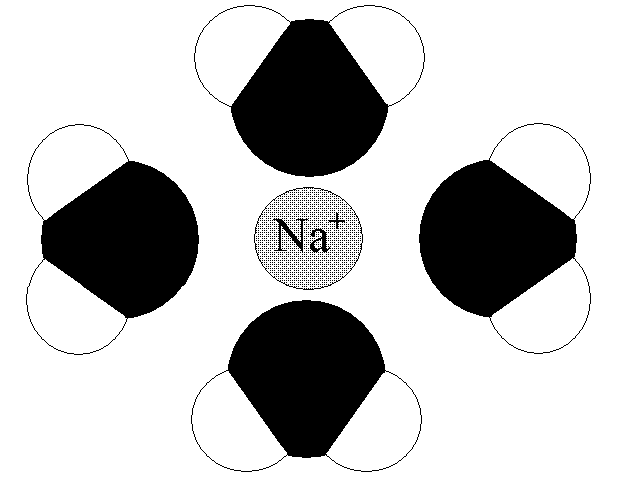
Figura 3: Idratazione dello ione sodio.
SOLUZIONI DI SOLUTI E SOLVENTI POLARI. Quando il soluto ed il solvente sono entrambi polari la sfera di solvatazione e' tenuta insieme da interazioni di tipo dipolo-dipolo. Un esempio di soluzione di questo tipo e' dato dalla coppia acqua-glucosio:
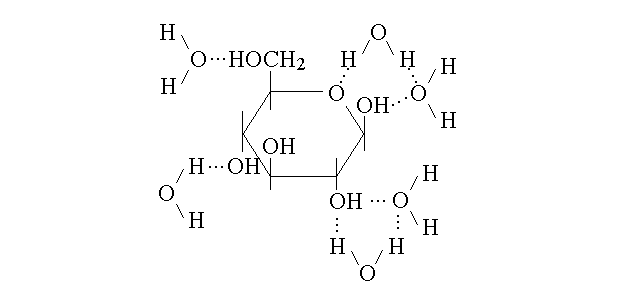
Figura 4: Idratazione del glucosio.
SOLUZIONI DEI GAS NEI LIQUIDI. Questo tipo di soluzione e' molto importante in medicina (si pensi agli scambi respiratori tra l'aria contenuta negli alveoli polmonari ed il sangue).
Se il gas non da reazioni chimiche con il liquido (ad es. ossigeno o azoto disciolti in acqua), la principale energia che governa la dissoluzione del gas e' l'entropia e non si formano affatto le sfere di idratazione. In questi casi il gas obbedisce alla legge di Henry, secondo la quale la concentrazione del gas e' direttamente proporzionale alla pressione parziale che esso esercita sulla superficie del liquido:
Il coefficiente di Henry (la k dell'eq. 15) e' diverso per ogni gas. Si definisce (un po' impropriamente) solubilita' del gas ad una certa pressione la concentrazione determinata secondo la legge di Henry quando il gas e' perfettamente in equilibrio con la soluzione.
Se invece il gas reagisce con il solvente, esso si trova in soluzione in varie forme, e la legge di Henry si applica soltanto alla forma che e' uguale negli stati si soluto e gassoso; tutte le altre forme sono "in piu'". In genere un gas di questo tipo risulta alquanto solubile (e certamente piu' solubile di uno che obbedisce alla legge di Henry). Ad esempio l'anidride carbonica (un altro gas importante per la respirazione) si trova in soluzione come:
CO2 + H2O <==> H2CO3 <==> HCO3- + H+
Quali forze guidano il dissolvimento del soluto nel solvente? Nel caso delle miscele dei gas ideali, le molecole semplicemente occupano lo stesso volume senza interagire le une con le altre, e sono soltanto mesoclate tra loro; l'energia che favorsice il mescolamento e' quindi di tipo entropico con nessun contributo entalpico (contributi entalpici diversi da zero, ma sempre modesti, possono aversi nel caso delle miscele di gas reali).
Anche nel caso delle soluzioni di gas in liquidi, i contributi entalpici sono trascurabili e il dissolvimento e' causato dall'aumento dell'entropia del sistema. Una soluzione di questo tipo, nella quale sono trascurabili i contributi entalpici e sono rilevanti soltanto quelli entropici e' chiamata una soluzione ideale.
Nel caso delle soluzioni di liquidi in liquidi o di solidi in liquidi si ha formazione delle sfere di solvatazione e si debbono considerare vari contributi rilevanti sia di tipo entalpico che di tipo entropico:
la formazione dei legami deboli tra soluto e solvente fornisce un contributo entalpico positivo (come sempre quando i legami vengono formati). Pero' perche' questo sia possibile e' necessario separare le molecole del soluto tra loro e quelle del solvente tra loro, rompendo i legami solvente-solvente e soluto-soluto; questo comporta un contributo entalpico negativo. Se l'energia rilasciata dai legami formati nella solvatazione supera quella assorbita dalla rottura dei legami tra le molecole del solvente e tra quelle del soluto, la dissoluzione del soluto e' esotermica (cioe' riscalda il sistema; un esempio e' dato dalla dissoluzione dell'idrossido di sodio in acqua). In caso contrario la dissoluzione del soluto nel solvente e' endotermica (e raffredda il sistema; un esempio e' dato dalla dissoluzione dell'urea in acqua). Si ricordi che l'entalpia di una reazione (in questo caso del dissolvimento) si manifesta sempre sotto forma di calore assorbito o ceduto (e quindi di variazione di temperatura del sistema).
La dispersione del soluto nel solvente comporta in genere un aumento di entropia del sistema (quindi e' favorita entropicamente, come accade nelle soluzioni ideali). Pero' la formazione delle sfere di idratazione ordina il solvente e quindi da un contributo entropico negativo (in genere inferiore al precedente).
Vi sono quattro proprieta' delle soluzioni che non dipendono dalla natura chimica del soluto ma soltanto dalla sua concentrazione; sono chiamate colligative e sono: la pressione osmotica, la variazione della pressione di vapore saturo, la variazione del punto di congelamento e la variazione del punto di ebollizione. Se il soluto e' un elettrolita e dissocia in ioni (oppure se dissocia per ragioni non elettrolitiche in particelle non cariche, caso piuttosto raro), ogni particella o ione si comporta come una molecola intera e la concentrazione dal punto di vista delle proprieta' colligative e' riferita alle particelle o ioni e non alle molecole indissociate. Per tenere conto di questo occorre impiegare il binomio di Van't Hoff.
CONCENTRAZIONE ANALITICA E CONCENTRAZIONE EFFETTIVA. e' una formula che consente di calcolare la concentrazione effettiva in particelle se sono note la concentrazione analitica molare del soluto e le caratteristiche della sua dissociazione. Definiamo:
Concentrazione molare analitica del soluto: C = n / V (con n = peso / P.M.)
Concentrazione molare effettiva del soluto: Ceff = neff / V
in pratica la concentrazione analitica e' quella che possiamo calcolare sapendo quanto soluto e' stato pesato sulla bilancia, e trascurando qualunque riferimento alla dissociazione; la concentrazione effettiva e' la concentrazione in particelle o ioni o molecole effettivamente presenti in soluzione. E' ovvio che neff > n e quindi Ceff > C.
CONCENTRAZIONE EFFETTIVA DEI SALI. I sali sono elettroliti forti, cioe' si disciolgono in acqua dissociandosi completamente negli ioni che li costituiscono (e non sono quindi presenti in soluzione nella forma delle molecole indissociate, ma soltanto degli ioni). Ad esempio: NaCl --> Na+ + Cl-. In soluzione non sono presenti molecole di NaCl (che del resto non sono presenti neppure nel cristallo di questo sale: infatti il reticolo alterna regolarmente ioni Na+ e ioni Cl-, e non consente di individuare vere molecole) ma solo ioni sodio e ioni cloruro. Si definisce con la lettera greca ν il numero di ioni che si originano dalla dissociazione di ogni unita' formale (molecola) di sale e si ottiene:
Ad esempio se noi disciogliamo una mole di cloruro di sodio in un litro d'acqua, noi otteniamo una mole di ione sodio e una mole di ione cloruro, e quindi: ν = 2; Ceff = 1 M x ν = 2 OsM (come sara' spiegato meglio piu' avanti l'osmolarita', abbreviata OsM, e' l'unita' di misura della concentrazione effettiva misurata in moli di particelle per litro di soluzione). Se invece sciogliamo in un litro d'acqua una mole di cloruro di calcio, che ha formula CaCl2 e dissocia interamente secondo la reazione CaCl2 --> Ca2+ + 2 Cl- (da una molecola si ottengono tre ioni), noi otteniamo : ν = 3; Ceff = 1 M x ν = 3 OsM.
DISSOCIAZIONE DEGLI ELETTROLITI DEBOLI. Gli elettroliti deboli sono sostanze che disciolte in acqua dissociano reversibilmente negli ioni che li costituiscono e quindi si trovano in soluzione sia nella forma di molecole indissociate che di ioni. Ad es.:
Si definisce grado di dissociazione, indicato con la lettera greca α la frazione:
E' evidente che α puo' assumere soltanto valori compresi tra zero (nessuna dissociazione) e 1 (dissociazione completa: tutte le molecole sono dissociate). Le molecole indissociate e dissociate sono rispettivamente:
Poiche' dalla dissociazione di ogni molecola che si dissocia originano ν particelle o ioni, le particelle originate dalle molecole dissociate risultano:
Sommando le molecole indissociate alle particelle originate dalla dissociazione delle molecole dissociate si ottiene:
Quest'ultima espressione costituisce il prodotto del numero di moli analitiche per il binomio di Van't Hoff, [1 + α (ν -1)].
FENOMENI OSMOTICI. Il fenomeno dell'osmosi si verifica quando due soluzioni dello stesso solvente a diversa concentrazione sono separate da una membrana semipermeabile (cioe' permeabile al solvente ma non al soluto; non importa che il soluto nelle due soluzioni sia uguale o diverso, purche' non possa attraversare la membrana). Questi fenomeni sono importanti per la fisiologia cellulare e vascolare perche' le membrane che delimitano le cellule e i vasi sono in genere semipermeabili o selettivamente permeabili (cioe' permeabili ad alcuni soluti ma non ad altri).
Quando si ha osmosi tra due soluzioni, la soluzione a concentrazione maggiore estrae solvente da quella a concentrazione minore:
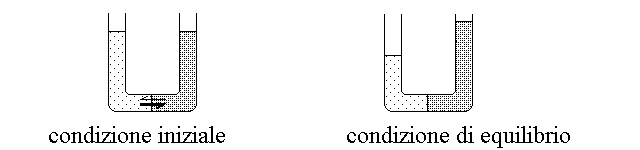
Attraverso una membrana semipermeabile c'e' sempre flusso di solvente nei due sensi; l'osmosi e' propriamente il flusso netto dalla soluzione piu' diluita verso quella piu' concentrata (cioe' la differenza tra i flussi in questa direzione e nella direzione opposta, il primo essendo sempre maggiore del secondo). Se si compie un esperimento di osmometria tra una soluzione ed il solvente puro, si puo' determinare la pressione osmotica della soluzione, cioe' la forza che attrae il solvente verso la soluzione. Se invece l'esperimento e' effettuato utilizzando due soluzioni, la forza che si misura e' pari alla differenza tra la pressione osmotica della soluzione piu' concentrata e quella della soluzione piu' diluita. L'esperimento dimostra che la pressione osmotica (che si indica con la lettera greca π) corrisponde a:
Questa equazione corrisponde esattamente all'equazione di stato dei gas perfetti ed R ha lo stesso valore. Per spiegare questo basta considerare che in un esperimento di osmometria condotto confrontando attraverso la membrana semipermeabile la soluzione ed il suo solvente puro, il soluto si espande come un gas perfetto, ma per aumentare il suo volume deve estrarre solvente attraverso la membrana.
MISURA DELLA PRESSIONE OSMOTICA. Per misurare la pressione osmotica di una soluzione e' necessario preparare un tubo ad U i cui due bracci siano separati dalla membrana semipermeabile, e porre in uno di essi la soluzione, nell'altro il solvente puro. A questo punto e' necessario esercitare sopra la soluzione, mediante un pistone, una pressione idrostatica tale da impedire il flusso osmotico. La pressione osmotica e' uguale nel modulo ma opposta nella direzione alla pressione idrostatica necessaria per impedire il flusso osmotico. Se lo stesso esperimento viene condotto utilizzando due soluzioni anziche una soluzione ed il solvente puro, la pressione idrostatica che e' necessario esrecitare sopra la soluzione a maggior concentrazione per impedire il flusso osmotico misura la differenza di pressione osmotica tra due soluzioni:
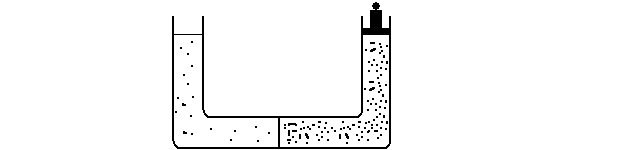
OSMOLARITA'. Quando e' stata definita la molarita' (M = n / V) si e' fatto riferimento al numero di moli analitico del soluto; per contro, nel caso delle proprieta' colligative, e segnatamente della pressione osmotica,si deve usare la molarita' in particelle, o molarita' effettiva. Si ricorre allora ad una unita' di misura omogenea con la molarita' definita osmolarita' (abbreviata OsM) e corrispondente al prodotto della molarita' per il binomio di Van't Hoff:
Si noti che nel caso dei sali e degli altri elettroliti forti si ha α = 1 e il binomio di Van't Hoff si riduce a ν.
PRESSIONE OSMOTICA DEI LIQUIDI BIOLOGICI. I liquidi biologici contengono molti soluti diversi e non tutte le mebrane dell'organismo sono impermeabili a tutti i soluti. Si descrivono quindi due tipi di pressione osmotica:
- La pressione osmotica vera e propria, che risulta dalla somma delle concentrazioni di tutti i soluti (tenendo conto delle rispettive dissociazioni ove necessario), e corrisponde a circa 7,6 atm alla temperatura di 37 C. Questo valore corrisponde ad una concnetrazione complessiva pari a circa 0,3 OsM (in genere non si possono sommare le molarita'; ma l'osmolarita' puo' risultare da una somma perche' la pressione osmotica e' colligativa e non dipende dalla natura chimica del soluto).
- La pressione colloido-osmotica (o oncotica) dovuta soltanto ai soluti di grande massa molare (in genere si considerano tali quelli con PM > 30.000), che corrisponde a circa 26 mmHg (0,035 atm) a 37 C.
Questa distinzione e' importante perche' alcune membrane, quali ad es. la membrana cellulare, sono pressoche' impermeabili a tutti o quasi tutti i soluti, mentre altre, quali quelle dei capillari sono impermeabili soltanto ai soluti a grande massa molecolare (e quindi non risentono della pressione osmotica dovuta ai soluti a basso peso molecolare).
FENOMENI OSMOTICI NEI VASI CAPILLARI. Il vaso capillare ha dei pori che lasciano passare l'acqua e i soluti a basso peso molecolare; la differenza di pressione osmotica tra il sangue all'interno del capillare e il liquido tissutale all'esterno dipende soltanto dalla diversa concentrazione dei soluti ad alto peso molecolare (ad es. le proteine del plasma).
All'estremita' arteriosa del capillare la pressione idrostatica (dovuta alla forza contrattile del cuore e alla dilatazione elastica delle grandi arterie) supera la perssione colloido-osmotica del sangue e forza il liquido ad uscire dal capillare per bagnare i tessuti; all'estremita' venosa avviene il contrario. Questo realizza un flusso di acqua e soluti a basso peso molecolare all'esterno del capillare, concentrico e parallelo al flusso ematico interno al capillare, il cui risultato e' quello di rimescolare il liquido tissutale:
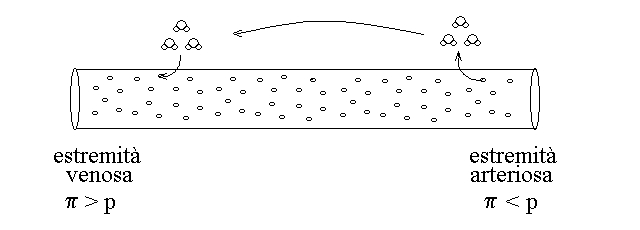
PRESSIONE OSMOTICA DEI LIQUIDI BIOLOGICI: SOLUZIONI ISOTONICHE. Una soluzione che ha la stessa pressione osmotica totale del sangue si definisce isotonica. Sono esempi di soluzioni isotoniche la soluzione di NaCl allo 0,9% peso/volume (soluzione fisiologica) o quella di glucosio al 5% peso/volume (soluzione glucosata); entrambe hanno concentrazione pari a 0,28 - 0,30 OsM e π = 7,6 atm a 37 C. Soluzioni a concentrazione maggiore sono definite ipertoniche, a concentrazione minore ipotoniche. Soltanto soluzioni isotoniche o molto prossime all'isotonicita' possono essere impiegate per l'infusione endovenosa: infatti le cellule ematiche confrontate con soluzioni ipotoniche si rigonfiano d'acqua fino a scoppiare (flusso osmotico dall'esterno verso l'interno della cellula); confrontate con soluzioni ipertoniche perdono acqua, si raggrinziscono e diventano non funzionali (questo danno e' in genere reversibile).
VARIAZIONE DELLA PRESSIONE DI VAPORE SATURO (LEGGE DI RAOULT). Ogni liquido e' in equilibrio con il suo vapore (o gas reale) che si produce costantemente alla superficie del liquido per evaporazione e che costantemente ricondensa sul liquido stesso. Si definisce pressione di vapore saturo la pressione del vapore in equilibrio con il liquido ad ogni determinata temperatura (infatti la pressione di vapore saturo aumenta con l'aumentare della temperatura). Ad esempio la pressione di vapore saturo dell'acqua e' 18 mmHg a 25 C e 760 mmHg a 100 C (temperatura di ebollizione).
Quando si discioglie un soluto in un solvente la pressione di vapore saturo della soluzione risulta intermedia tra quella del solvente e quella del soluto. La pressione di vapore saturo della soluzione puo' essere calcolata con una equazione dovuta a F.M. Raoult, purche' siano note le frazioni molari di ogni componente della soluzione e le corrispondenti pressioni di vapore saturo:
In questa equazione il componente 1 e' il solvente e i successivi i diversi soluti presenti (che possono essere uno o piu'; per gli elettroliti si deve contare separatamente ogni specie ionica). Se il soluto e' solido (ad es. NaCl o glucosio), la sua pressione di vapor saturo e' molto piccola e puo' essere trascurata; la legge di Raoult si semplifica in:
Poiche' la frazione molare (X) di qualunque componente di una miscela e' inferiore all'unita', la pressione di vapore saturo della soluzione in questo caso risulta inferiore a quella del solvente puro (cioe' intermedia tra quella del solvente e quella del soluto che in questa approssimazione e' stata assunta uguale a zero). Un'altra forma della legge di Raoult per il soluto non volatile, analoga alla precedente e':
La distillazione e' una procedura finalizzata a separare il soluto dal solvente, o quanto meno ad arricchire il soluto rispetto al solvente (aumentare la concentrazione) e costituisce una applicazione fondamentale della legge di Raoult. Quando il soluto ed il solvente hanno differenti pressioni di vapore saturo, la soluzione bolle alla temperatura alla quale la somma delle pressioni di vapore saturo dei due componenti eguaglia la pressione atmosferica, e la fase di vapore risulta arricchita del componente piu' volatile. facendo raffreddare e liquefare il vapore si ottiene una nuova soluzione nella quale risulta aumentata la concentrazione del componente più volatile e diminuita quella del componente meno volatile, come dallo schema seguente.
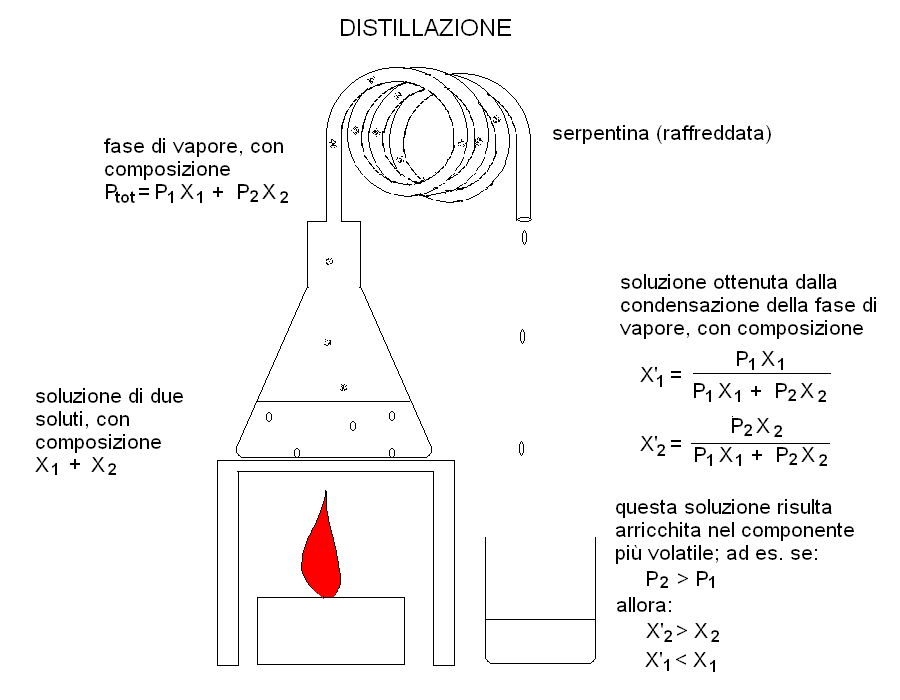
Siano X1 e X2 le frazioni molari dei due componenti di una soluzione e P1 e P2 le rispettive pressioni di vapor saturo alla temperatura di ebollizione della soluzione. La frazione molare dei due componenti in fase di vapore risultera':
X'1 = P1X1 / (P1X1 + P2X2)
X'2 = P2X2 / (P1X1 + P2X2)
Queste equazioni non si prestano ad ovvie semplificazioni e non sono facili da visualizzare; ciononostante e' possibile considerare un esempi per illustrare il funzionamento del sistema di distillazione. Se P2 = 5 P1 e X2 = 0.1 (con X1 = 0.9) nella fase gassosa si avra' X'2 = 0.357 (con X'1 = 0.643). Se il liquido ottenuto dalla condensazione del vapore viene ridistillato si ottiene X"2 = 0.735 (con X"1 = 0.265). Si noti che questo calcolo semplificato applica alla distillazione di una piccola parte della soluzione iniziale, tale che le frazioni molari della soluzione si possano considerare invariate. L'arricchimento di X2 e' pari a 3,57 volte nella prima distillazione e di ulteriori 2,06 volte nella seconda distillazione (al termine della seconda distillazione l'arricchimento complessivo di X2 risulta pari a 3,57x2,06 = 7,35 volte).
VARIAZIONE DEL PUNTO DI GELO E DEL PUNTO DI EBOLLIZIONE. Sono entrambe conseguenze della variazione della pressione del vapore saturo, ma si calcolano con riferimento alla molalita' effettiva (uguale alla molalita' analitica moltiplicata per il binomio di Van't Hoff) perche' richiedono temperature molto diverse e la molalita' ha il vantaggio di essere indipendente dalla temperatura. Le leggi (per il caso del soluto non volatile) sono:
Dove Kcr rappresenta la costante crioscopica (di congelamento; -1,86 gradi/m per l'acqua) e Keb la costante ebullioscopica (di ebollizione; +0,52 gradi/m per l'acqua) caratteristiche entrambe del solvente e non del soluto.
Torna a: didattica; pagina iniziale.