PRIMA FACOLTA' DI MEDICINA E CHIRURGIA - CLUPS DIETISTA; INFERMIERE; TECNICO DI LABORATORIO
SAPIENZA UNIVERSITA' DI ROMA
Gli alimenti ingeriti con la dieta devono essere DIGERITI nell'intestino (cioe' scomposti nei loro costituenti a minor peso molecolare) prima di essere ASSORBITI (trasferiti dal lume intestinale al sangue). I processi digestivi sono dovuti ad enzimi specializzati prodotti dalle ghiandole esocrine annesse all'intestino (gh. salivari, cellule specializzate della mucosa gastrica, pancreas). La digestione degli alimenti assolve due funzioni: (1) in primo luogo l'organismo non puo' usare le macromolecole esterne introdotte con la dieta, ma deve sintetizzare le proprie (si vedano gli argomenti sintesi proteica e metabolismo), a partire dai monomeri che le costituiscono; i processi digestivi assolvono questa funzione convertendo macromolecole in monomeri. (2) Le macromolecole assunte con la dieta potrebbero essere tossiche, mentre i monomeri che le costituiscono non lo sono; inoltre i microorganismi che inevitabilmente contaminano gli alimenti potrebbero essere patogeni; i processi digestivi hanno quindi anche la funzione di proteggere l'organismo.
Poiche' i processi digestivi differiscono a seconda del tipo di molecola che era contenuta nell'alimento ingerito (ogni alimento contiene in proporzioni variabili acqua, sali, glicidi, lipidi e proteine; in minor misura acidi nucleici e altri componenti), e' conveniente descrivere separatamente i processi digestivi a carico di ciascuna classe di macromolecole biologiche.
LA DIGESTIONE DEI GLICIDI. I glicidi sono presenti negli alimenti in forma di polisaccaridi (principalmente l'amido degli alimenti di origine vegetale), disaccaridi (principalmente saccarosio nella frutta e lattosio nel latte e nei suoi derivati) e, piu' raramente, monosaccaridi (ad es. il glucosio in alcuni frutti e il fruttosio nel miele).
La digestione dell'amido e del glicogeno e' dovuta alle AMILASI (enzimi prodotti dalle ghiandole salivari e dal pancreas) che degradano il polimero producendo frammenti progressivamente sempre piu' piccoli chiamati maltodestrine. I prodotti finali di questo processo sono i disaccaridi maltosio e isomaltosio, le cui formule sono riportate in una lezione precedente.
I disaccaridi non possono essere assorbiti come tali dalle cellule intestinali ma subiscono una ulteriore digestione che li idrolizza a monosaccaridi; gli enzimi responsabili sono le disaccaridasi. Poiche' l'uomo possiede solo quattro disaccaridasi, soltanto quattro disaccaridi possono essere digeriti a monosaccaridi dal nostro intestino:
1) l'enzima maltasi idrolizza il maltosio trasformandolo in due molecole di glucosio.
2) L'isomaltasi idrolizza l'isomaltosio trasformandolo in due molecole di glucosio.
3) La lattasi idrolizza il lattosio trasformandolo in una molecola di glucosio e una di galattosio.
4) La saccarasi idrolizza il saccarosio trasformandolo in una molecola di glucosio e una di fruttosio.
I monosaccaridi possono essere assorbiti dalle cellule intestinali, che li riversano nel sangue del territorio mesenterico-portale e tramite questo raggiungono il fegato che puo' conservarli sotto forma di glicogeno. I polisaccaridi e disaccaridi che introduciamo con la dieta ma che non possiamo digerire per la mancanza degli enzimi necessari non possono essere utilizzati e finiscono nelle feci sotto forma di "fibra"; la principale sorgente di fibra della dieta e' la cellulosa (un omopolimero del glucosio diverso dall'amido).
LA DIGESTIONE DEI TRIGLICERIDI. I principali lipidi della dieta sono i trigliceridi contenuti negli oli vegetali e nei grassi di origine animale. La digestione dei trigliceridi avviene ad opera di enzimi chiamati lipasi che idrolizzano il trigliceride ad un monogliceride e due acidi grassi; questi componenti vengono separatamente assorbiti dalle cellule intestinali che li utilizzano per risintetizzare trigliceridi da rilasciare infine nel sangue a livello del circolo mesenterico-portale sotto forma di chilomicroni (gocciole microscopiche di grassi).
LA DIGESTIONE DELLE PROTEINE. La digestione delle proteine avviene ad opera di enzimi chiamati endopeptidasi (proteasi) ed esopeptidasi, essenzialmente in due sedi: lo stomaco ed l'intestino tenue. La mucosa dello stomaco secerne una proteasi principale, la PEPSINA, e alcune proteasi accessorie specifiche per proteine particolari (ad es. la chimosina o rennina, specifica per la digestione della caseina del latte). Questi enzimi frammentano le proteine in polipeptidi a peso molecolare piu' basso e funzionano a pH fortemente acido; infatti l'acidita' del succo gastrico svolge la funzione essenziale di denaturare le proteine rendendole piu' sensibili alla digestione (e inoltre ha una efficace azione battericida). I polipeptidi cosi' prodotti passano dallo stomaco al duodeno dove incontrano altre endopeptidasi prodotte dal pancreas (TRIPSINA, CHIMOTRIPSINA, elastasi ed altre ancora) che li frammentano ulteriormente. I piccoli polipeptidi cosi' prodotti vengono infine idrolizzati a singoli aminoacidi dalle esopeptidasi (chiamati aminopeptidasi se idrolizzano il polipeptide a partire dall'estremita' -NH2, carbossipeptidasi se lo idrolizzano a partire dall'estremita' -COOH). Le aminopeptidasi sono associate alla membrana delle cellule intestinali; le carbossipeptidasi sono presenti nel succo pancreatico. I singoli aminoacidi prodotti alla fine del processo digestivo vengono infine assorbiti dalle cellule intestinali.
FENOMENI PATOLOGICI A CARICO DEI PROCESSI DIGESTIVI: ALLERGIE E INTOLLERANZE. Si chiamano intolleranze alimentari le condizioni, spesso ereditarie, per cui l'ingestione di un alimento o di un gruppo di alimenti causa sintomi intestinali dovuti all'incapacita' dell'organismo di digerirlo ed assorbirlo. L'alimento indigerito costituisce nutrimento per la flora batterica e puo' favorire l'insorgenza di enteriti batteriche. La causa piu' frequente di intolleranze alimentari e' la carenza di uno specifico enzima digestivo. Ad esempio in molte etnie umane gli adulti smettono di produrre la lattasi e diventano intolleranti agli alimenti che contengono lattosio. Questa condizione non e' grave ma richiede che l'eliminazione del latte e dei suoi derivati dalla dieta, o almeno una loro drastica riduzione. E' molto grave invece la carenza genetica di lattasi nel neonato, che impone la sostituzione del latte materno con un latte artificiale nel quale il lattosio e' stato predigerito o sostituito con glucosio.
Esistono malattie metaboliche nelle quali non ci sono difetti nella digestione e assorbimento dei nutrienti, ma nel loro metabolismo successivo: ad esempio la fenilchetonuria e' un difetto nel catabolismo degli aminoacidi fenilalanina e tirosina, che se ingeriti in eccesso risultano tossici. La condizione e' presente fin dalla nascita e il neonato deve essere alimentato con cibi artificialmente impoveriti di questi aminoacidi. Una dieta normale infatti gli causerebbe danni cerebrali gravi e ritardo nello sviluppo intellettuale. Queste condizioni non sono da tutti considerate tra le intolleranze alimentari perche' la sintomatologia non e' a carico degli organi digerenti, e sono in genere considerate tra i dismetabolismi. Un caso interessante e peculiare e' quello del favismo. In questa malattia genetica, ereditaria, il paziente presenta una variante dell'enzima glucosio 6 fosfato deidrogenasi che viene inibita da una sostanza presente nelle leguminose (soprattutto nelle fave). Non solo l'ingestione di questi legumi, ma anche l'inalazione del loro polline genera un quadro dismetabolico molto grave perche' blocca la via dei pentoso fosfati.-----
Molto diverso e' il caso delle allergie alimentari. Le allergie sono malattie nelle quali l'organismo produce anticorpi della classe delle IgE contro vari antigeni; forme allergiche tipiche sono le pollinosi, le riniti e la c.d. febbre da fieno. Gli anticorpi in genere hanno funzione difensiva, ma nelle malattie allergiche sono prodotti contro sostanze innocue e scatenano reazioni difensive esagerate, inutili e dannose. Nel caso delle allergie alimentari gli antigeni contro i quali il corpo reagisce sono presenti negli alimenti. Sono condizioni acquisite (non presenti alla nascita) e potenzialmente gravi, occasionalmente letali. Ovviamente il malato deve evitare in modo assoluto i cibi ai quali e' allergico ed e' consigliabile effettuare prove cutanee di sensibilita' agli allergeni.
Alcune malattie presentano quadri misti, sia di intolleranza che di allergia: ad esempio nel morbo celiaco il paziente ha una intolleranza alla gliadina, una proteina contenuta nel grano e nelle farine che se ne producono. I frammenti parzialmente digeriti e non assorbiti della proteina si comportano come allergeni e generano risposte allergiche. Anche questa malattia puo' essere alquanto grave.
Molti microorganismi e tutte le piante capaci di fotosintesi sono in grado di produrre da se stessi tutti i componenti organici necessari al loro metabolismo, a partire da una sorgente qualsiasi di carbonio (ad es. glucosio; CO2 nel caso delle piante capaci di fotosintesi), di azoto (amine o ammidi) e da acido fosforico. Gli animali invece necessitano di introdurre con la dieta anche alcuni composti organici che sono necessari al metabolismo ma che non vengono sintetizzati dal loro organismo; questi composti sono chiamati ESSENZIALI. I nutrienti essenziali per l'uomo sono:
1) le vitamine.
2) Gli 8 aminacidi essenziali: fenilalanina/tirosina; isoleucina; leucina; lisina; metionina/cisteina; treonina; triptofano e valina. Inoltre l'arginina che sfugge al ciclo dell'urea (nel quale e' prodotta ma anche degradata) e' insufficiente al fabbisogno e deve quindi essere fornita con la dieta. Gli aminoacidi che compaiono in coppia nell'elenco possono essere convertiti gli uni negli altri ma non prodotti da altre fonti (ad es. e' possibile convertire fenilalanina in tirosina ma almeno uno dei due deve essere fornito con la dieta).
3) Gli acidi grassi poliinsaturi (ad es. l'acido arachidonico, necessario per la biosintesi delle prostaglandine) e alcuni composti analoghi (ad es. l'acido lipoico).
LE VITAMINE. Le vitamine sono piccole molecole organiche delle quali il nostro organismo ha bisogno ma che non puo' sintetizzare (o almeno non puo' sintetizzare a partire da precursori semplici), e che devono essere contenute nella dieta. La scoperta che la dieta deve contenere sostanze particolari, pena lo sviluppo di malattie specifiche (le avitaminosi o carenze vitamiche), e' abbastanza antica, ma la caratterizzazione chimica delle vitamine e' incominciata intorno nel 1912 con i lavori di Casimir Funk che isolo' la vitamina B1. Le vitamine sono convenzionalmente divise in liposolubili (prevalentemente contenute in alimenti grassi) e non liposolubili.
| | ||||
| vitamina | contenuta in | fabbisogno giornaliero | ruolo fisiologico | malattia da carenza |
| A (retinolo) | vegetali (carote) | 0,75 mg | sviluppo dei tessuti osseo ed epiteliale; cofattore delle proteine della retina (visione) | difetti della visione; danni a carico della cute e delle mucose |
| D (calciferolo) | grassi animali e vegetali (prodotta anche dall'irraggiamento solare dei grassi della cute) | (0,01 mg) | assorbimento del calcio | rachitismo (nel bambino); osteomalacia (nell'adulto) |
| E (tocoferolo) | vegetali | 10-20 mg | antiossidante | |
| K (menadione) | prodotta dalla flora intestinale | (0,1 mg) | necessaria per la produzione degli enzimi della coagulazione | (sindromi emorragiche) |
| | ||||
| B1 (tiamina) | cuticola dei cereali | 1,5 mg | precursore del coenzima delle decarbossilasi | Beri-beri e malattia di Wernicke-Korsakoff (due diverse malattie neurologiche) |
| B2 (riboflavina) | molto diffusa | 1.5 mg | precursore dei coenzimi FAD e FMN | |
| B3 (niacina) | lievito, carni | 20 mg | precursore dei coenzimi NAD+ e NADP+ | pellagra (malattia della cute e del sistema nervoso) |
| B6 (pridossina) | molto diffusa | 2 mg | precursore del coenzima delle transaminasi | |
| B12 (cobalamina) | fegato, cereali, molluschi (l'assorbimento richiede una specifica proteina prodotta dallo stomaco) | 0,003 mg | coenzima di alcune metilasi/demetilasi | Anemia perniciosa (anemia megaloblastica e sintomi neurologici) |
| acido folico | molto diffusa | 0,4 mg | coenzima di alcune metilasi/demetilasi | Anemia megaloblastica |
| acido folico | molto diffusa | 0,4 mg | coenzima di alcune metilasi/demetilasi | Anemia megaloblastica |
| acido lipoico | molto diffusa | ? | coenzima di alcune ossidoreduttasi | |
| acido pantotenico | molto diffusa | 10 mg | precursore del coenzima A | |
| C (acido ascorbico) | agrumi | 100 mg | antiossidante | Scorbuto |
| H (biotina) | molto diffusa | 0,03 mg | coinvolta nelle reazioni di decarbossilazione | |
Il sangue puo' essere frazionato in due componenti essenziali: la parte corpuscolata che include i globuli rossi, i bianchi e le piastrine, e la parte liquida che si chiama plasma. Se si permette la coagulazione dopo il prelievo, il plasma viene deprivato del fibrinogeno, che precipita sotto forma di fibrina, e cio' che ne rimane prende il nome di siero. Il siero del sangue contiene fisiologicamente alcuni enzimi necessari alle sue funzioni, quali ad es. quelli della coagulazione; contiene inoltre enzimi che dovrebbero essere intracellulari ma che si liberano nel siero in conseguenza della morte delle cellule che li contengono. L'indagine diagnostica piu' semplice che puo' essere condotta sulla parte corpuscolata del sangue e' l'esame emocromocitometrico, che consiste nel separare e contare i vari tipi cellulari (al microscopio o con strumenti automatizzati). Per gli studi del siero invece si utilizza l'elettroforesi che consiste nel deporre una goccia di siero su un supporto (in genere un particolare tipo di carta assorbente) e far muovere le proteine in un campo elettrico; questo separa le proteine in base alla loro carica elettrica. Per rendere visibili le proteine si adoperano coloranti specifici. Le macchie colorate cosi' ottenute possono essere lette da uno strumento (riflettometro) che produce un grafico chiamato protidogramma:
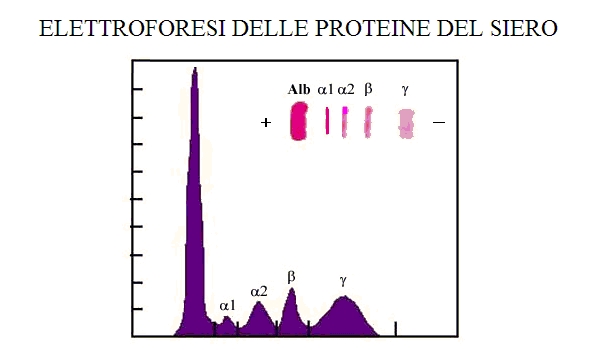
La concentrazione delle proteine del plasma e' di circa 7 g/100 mL, e la proteina piu' abbondante nel protidogramma e' l'albumina che da sola costituisce circa il 40% delle proteine totali. L'albumina forma una intensa banda colorata verso il polo positivo ed e' la proteina del siero con maggiore carica negativa (per questo si muove piu' rapidamente delle altre verso il polo positivo. Dietro l'albumina migrano nel campo elettrico le globuline alfa, poi le beta ed infine le gamma.
L'EMOGLOBINA
L'emoglobina e' una proteina del sangue contenuta all'interno dei globuli rossi ma non nel plasma: pertanto non appare nel protidogramma. La sua concentrazione, misurata nel sangue intero, e' molto elevata, pari a circa 14 g/100 mL. E' costituita da quattro subunita': due catene polipeptidiche chiamate alfa e due beta; ciascuna di queste porta una molecola organica piuttosto complessa chiamata eme che ha al centro uno ione Fe+2. L'eme ha un colore intensamente rosso e l'emoglobina e' responsabile del colore del nostro sangue. La funzione dell'emoglobina e' il trasporto dell'ossigeno, che si combina reversibilmente con il ferro dell'eme e puo' essere assorbito dal sangue nei capillari polmonari e ceduto ai tessuti. Si deve ricordare che, come tutti i gas che seguono la legge di Henry, l'ossigeno e' poco solubile in acqua e la sua concentrazione nel sangue e' pari a circa 0,2 mM; l'emoglobina invece e' solubile e molto abbondante e la sua "concentrazione" nel sangue (ignorando il problema della sua distribuzione soltanto all'interno dei globuli rossi) e' poco meno di 2,5 mM. Se si considera che ogni molecola di emoglobina contiene quattro emi e quindi si combina con quattro molecole di ossigeno, si conclude che l'ossigeno puo' essere contenuto nel sanuge arterioso nella misura di poco meno di 10 mMoli / L, e che soltanto il 2% di esso e' fisicamente disciolto nei liquidi ematici mentre il 98% e' legato all'emoglobina.
LA COAGULAZIONE DEL SANGUE.
La coagulazione del sangue e' un processo biochimico complesso che avviene in due fasi. La prima risposta alla lesione di un vaso sanguigno (che puo' aprirsi sia in una ferita esterna che in un sanguinamento interno) e' l'AGGREGAZIONE DELLE PIASTRINE, corpuscoli subcellulari presenti nel sangue in numero di circa 200.000/mmc. Il tappo piastrinico cosi' formato arresta l'emorragia ma e' instabile e dura soltanto poche ore; ha lo scopo di permettere la FORMAZIONE DELLA RETE DI FIBRINA, che costituisce la seconda fase del processo di coagulazione. La fibrina e' una proteina che viene deriva dalla degradazione di una proteina circolante prodotta dal fegato e chiamata FIBRINOGENO. Il fibrinogeno e' solubile grazie al fatto che alle estremita' della catena polipeptidica sono presenti residui aminoacidici carichi e molto idrofilici; una proteasi specifica chiamata TROMBINA ha la funzione di tagliare via queste estremita' (i fibrinopeptidi): il prodotto e' una proteina piu' piccola, la fibrina, insolubile che precipita e forma una rete macromolecolare sul tappo piastrinico. Poiche' la trombina deve convertire il fibrinogeno in fibrina soltanto in presenza di una emorragia, essa circola nel sangue nella forma di un precursore inattivo, la PROTROMBINA, che a sua volta deve essere attivato mediante taglio proteolitico.
Il controllo biochimico dell'attivazione della protrombina e' piuttosto complesso perche', per permettere la coagulazione rapida in caso di bisogno il sangue contiene tutte le proteine necessarie in forma inattiva, ma deve essere evitato il rischio di una loro attivazione non necessaria. Quando il meccanismo di controllo fallisce si hanno sindromi cliniche piuttosto gravi come la trombosi o la coagulazione intravascolare disseminata. Il controllo dell'attivazione della protrombina e' affidato ad una cascata di proteasi, chiamate i fattori della coagulazione, ciascuna delle quali e' presente in forma inattiva e viene attivata dal fattore che la precede nella cascata; una volta attivata ha la capacita' di attivare il fattore successivo.
L'attivazione della coagulazione segue due strade, chiamate intrinseca ed estrinseca. Nella via intrinseca il contatto del sangue con pareti diverse dall'endotelio vascolare causa la conversione proteolitica del fattore XII in fattore XII attivato (XIIa). Il fattore XIIa e' una proteasi specifica che converte il fattore XI in XIa e questo a sua volta il IX in IXa. Il fattore IX attivato (IXa) ed il fattore VIII attivato (VIIIa) agendo insieme attivano il fattore X il quale finalmente e' responsabile di convertire la protrombina in trombina. Una catena complessa come questa protegge l'organismo dalla coagulazione intravascolare accidentale perche' e' improbabile che si attivino insieme, in assenza di una lesione dell'endotelio, sia il fattore IX che il fattore XIII. In cambio, purtroppo, a causa di difetti genetici ereditari, e' possibile che l'attivazione della protrombina sia inefficiente: il difetto del fattore VIII costituisce l'emofilia classica (emofilia A), mentre il difetto del fattore IX ne costituisce una variante (emofilia B).
La via estrinseca richiede anch'essa due fattori attivati: il fattore VII e il fattore tissutale (tromboplastina); questi due fattori, agendo insieme, attivano il fattore X il quale, come nella via intrinseca e' l'attivatore della protrombina.
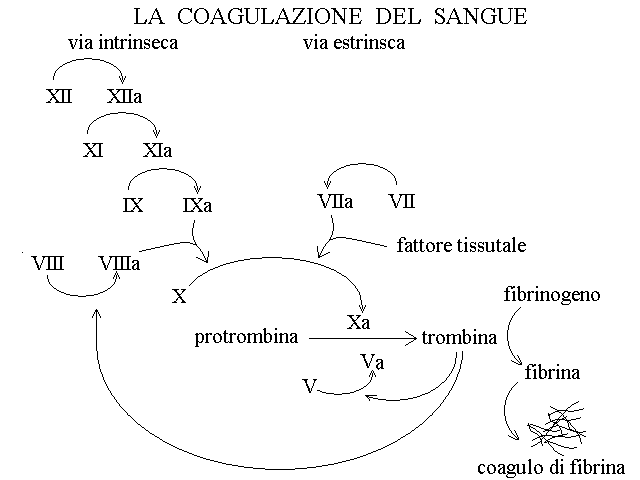
Che differenza c'e tra i fattori della coagulazione e le loro controparti attivate, ad esempio tra la protrombina e la trombina o tra il fattore X e l'Xa? Per fortuna il meccanismo di attivazione dei fattori della coagulazione e' piuttosto stereotipato. Ogni fattore della coagulazione e' una proteasi il cui sito attivo e' occupato da una "coda" della stessa catena polipeptidica; quando la coda viene digerita dal fattore precedente (ad es. la coda del fattore X viene tagliato dal fattore IXa), il sito attivo si libera e e diventa capace di attivare il fattore successivo (ad es. il fattore Xa taglia la coda di catena polipeptidica che occupa il sito attivo della trombina).
GLI ANTICORPI
Gli anticorpi sono proteine prodotte e secrete nel sangue dai linfociti B o dalle plasmacellule (che derivano dai linfociti B). Sono chiamati anche immunoglobuline (Ig) e sono di vari tipi: IgG, IgE, IgM, IgA. La struttura piu' tipica e' quella delle IgG, costituite da quattro catene polipeptidiche, due leggere (L) e due pesanti (H), legate tra loro da ponti disolfuro tra residui di cisteina. Ogni molecola di IgG ha la forma di una Y e alle due estremita' costituite da una catena L e una catena H presenta una regione che e' capace di combinarsi con altre molecole (chiamate antigeni). Ogni IgG presenta quindi due siti di legame per l'antigene, tra loro identici:
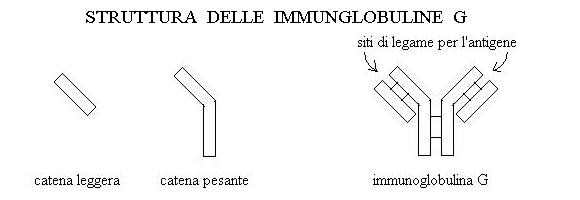
Le IgE hanno struttura simile alle IgG e sono responsabili in particolare di alcune malattie allergiche; le IgM sono costituite da cinque molecole, ciascuna simile ad una IgG, legate tra loro; le IgA che al contrario delle precedenti non si trovano nel sangue ma sono caratteristiche delle secrezioni mucose sono costituite da tre molecole, ciascuna simile ad una IgG, legate tra loro.
La funzione delle immunoglobuline e' quella di combinarsi con sostanze o organismi estranei eventualmente penetrati nel sangue e neutralizzarli: sono quindi fondamentali per la difesa dell'organismo in corso di infezioni virali, batteriche e parassitarie. I linfociti B richiedono un certo tempo per produrre le immunoglobuline adatte a neutralizzare un microorganismo che incontrano per la prima volta, quindi a seguito di una infezione virale o batterica, l'organismo umano si trova inizialmente poco difeso. Nello spazio di due o tre settimane appaiono nel sangue prima le IgM e poi le IgG specifiche per il microorganismo invasore e questo in genere avvia il processo di guarigione dall'infezione. Se lo stesso microorganismo attacca nuovamente l'individuo, la produzione di anticorpi e' precoce perche' i linfociti B conservano una "memoria" della precedente infezione e la malattia non si sviluppa. Questa e' la ragione per la quale alcune malattie, quali ad es. il morbillo o la parotite epidemica (orecchioni) ci colpiscono una sola volta nella vita; e' anche la ragione per la quale se l'organismo viene sottoposto ad infezione con una variante attenuata del germe (vaccinazione) diventa immune dalla malattia e non se ne ammala mai. Purtroppo non tutte le malattie infettive danno memoria e immunita', per varie ragioni, e quindi di alcune possiamo ammalarci piu' volte nella vita.
Nell'elettroforesi del siero le immunoglobuline migrano nella frazione delle gamma globuline.
ENZIMI PRESENTI NEL SANGUE NEL CORSO DI MALATTIE
Il sangue contiene enzimi che dovrebbero essere intracellulari ma che si liberano nel siero in conseguenza della morte delle cellule che li contengono; il loro dosaggio ha interesse diagnostico. I principali tra questi sono:
TRANSAMINASI: nel sangue si trovano nel sangue a bassa concentrazione due enzimi che scambiano tra loro gruppi aminici e chetonici: la glutamico-ossalacetico transaminasi (GOT o AST) e la glutamico-piruvico transaminasi (GPT o ALT). La reazione catalizzata dalla GPT, qui riportata a titolo di esempio, e' la seguente:
CH3-CO-COOH + COOH-(CH2)2-CHNH2-COOH <==> CH3-CHNH2-COOH + COOH-(CH2)2-CO-COOH
Questi enzimi derivano principalmente dal ricambio delle cellule del fegato, che in piccolissima percentuale, ogni tanto, muoiono e vengono sostituite: le cellule morte rilasciano il loro contenuto enzimatico (quindi anche la GPT e la GPT) nel sangue. Nel corso di malattie che causano la morte di cellule epatiche in gran numero, quali ad esempio le epatiti virali, le transaminasi epatiche vengono liberate nel sangue in grande quantita'; quindi un signficativo aumento della GOT e della GPT e' diagnostico per una malattia epatica.
CREATINA FOSFOCHINASI (CPK) E CITOCROMO c: sono enzimi caratteristici delle cellule muscolari e cardiache; la loro concentrazione ematica aumenta in corso di infarto del miocardio o di malattie della muscolatura scheletrica.
LATTICO DEIDROGENASI (LDH): un enzima presente in molti tessuti la cui concentrazione ematica aumenta nel corso di patologie muscolari e cardiache, del sistema nervoso centrale, del fegato e del tessuto ematopoietico.
Gli ormoni sono molecole di vario tipo prodotte da organi specializzati chiamati ghiandole endocrine e riversati nel sangue. Svolgono la funzione di regolatori di varie funzioni fisiologiche: cioe' le cellule dell'organismo in presenza di ogni ormone alterano in modo specifico la loro funzione. Dal punto di vista biochimico i principali ormoni possono essere: (1) piccole proteine; (2) derivati di aminoacidi; (3) derivati del colesterolo. Gli ormoni solubili in acqua (tipicamente quelli proteici) non hanno difficolta' a viaggiare nel sangue ma non possono penetrare attraverso la membrana cellulare e rimangono al di fuori della cellula; si combinano con una specifica prteina di membrana della cellula bersaglio (recettore) ed e' quest'ultima a trasmettere il segnale all'interno della cellula, in genere mediante un "secondo messaggero" (che puo' essere lo ione calcio o una molecola sintetizzata allo scopo come l'amp ciclico, cAMP). Gli ormoni non solubili in acqua sono trasportati nel sangue legati a proteine (in genere all'albumina); pero' attraversano la membrana cellulare ed in genere raggiungono il nucleo cellulare dove trovano il loro recettore e partecipano alla regolazione dell'espressione genica.
| | ||||
| ormone | prodotto da | organi bersaglio | ruolo fisiologico | malattie correlate |
| Somatotropina (GH) (piccola proteina) |
Ipofisi anteriore (adenoipofisi) | tutti i tessuti dell'organismo | Stimolo della replicazione cellulare e dell'accrescimento | Eccesso di secrezione: gigantismo o acromegalia; difetto di secrezione: nanismo armonico |
| Adrenocorticotropina (ACTH) (piccola proteina) |
Ipofisi anteriore (adenoipofisi) | corteccia surrenali | Stimolo della secrezione di degli ormoni glicocorticoidi, mineralcorticoidi e sessuali | Eccesso di secrezione: morbo di Cushing difetto di secrezione: morbo di Addison |
| Tireotropina (GH) (piccola proteina) |
Ipofisi anteriore (adenoipofisi) | Tiroide | Stimolo della secrezione di T3 e T4 | Eccesso di secrezione: ipertiroidismo; difetto di secrezione: ipotiroidismo |
| Tetra-Iodo-Tironina (tiroxina, T4) e Tri-Iodo-Tironina (T3) (derivati dell'aminoacido tirosina) |
Tiroide | tutti i tessuti dell'organismo | disaccoppiamento della fosforilazione ossidativa; produzione di calore, aumento del metabolismo | Eccesso di secrezione: ipertiroidismo; difetto di secrezione: ipotiroidismo |
| Tireocalcitonina (piccola proteina) |
Tiroide | osso, intestino, rene | promuove la deposizione di calcio nelle ossa | |
| Paratormone (piccola proteina) |
Paratiroidi | osso, intestino, rene | promuove il riassorbimento di calcio dall'osso e ne regola l'assorbimento e l'eliminazione | Eccesso di secrezione: osteoporosi |
| Cortisolo e altri glicocorticoidi (derivati del colesterolo) |
Corteccia surrenale | tutti i tessuti dell'organismo | Eccesso di secrezione: morbo di Cushing difetto di secrezione: morbo di Addison | |
| Aldosterone e altri mineralcorticoidi (derivati del colesterolo) |
Corteccia surrenale | rene | promuove il riassorbimento di ione potassio dalle urine | |
| Adrenalina e nor-adrenalina (derivati dell'aminoacido tirosina) |
Midolla surrenale | Cuore, muscolatura della parete arteriosa | aumento della frequenza e dell'energia della contrazione cardiaca; aumento della pressione arteriosa | Eccesso di secrezione (feocromocitoma): crisi ipertensive e tachicardia |
Torna a: didattica; pagina iniziale.