Course of LABORATORY MEDICINE
Hemostasis and Thrombosis
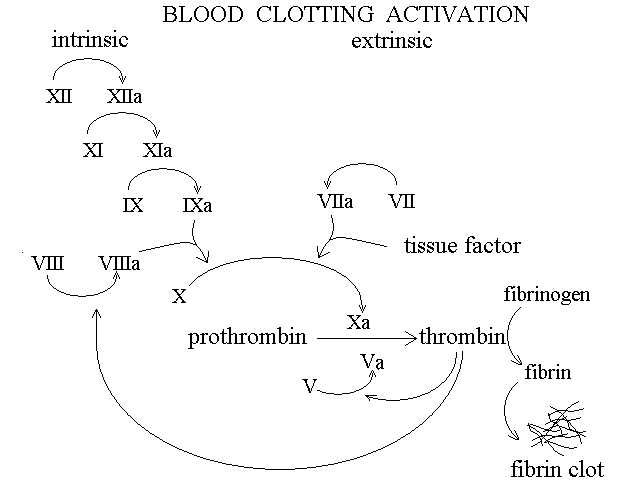
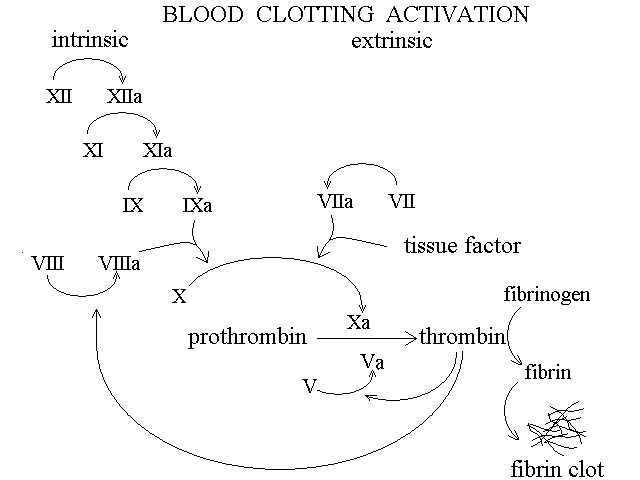
Hemostasis (blood coagulation; blood clotting) is the physiological process that stops an hemorrage and forms the semi-solid blood clot, which is then reabsorbed in the course of several days, in parallel with tissue repair. Hemostasis is actually costituted by two independent and very different processes: platelet aggregation and fibrin precipitation. Platelet aggregation is faster but the platelet plug is short lived; fibrin precipitation is slower but yields the much longer lived fibrin clot. As any other phyiological process, blood coagulation may go wrong because of many reasons and several pathologies related to abnormal coagulation are known.
Platelet activation. Platelets (thrombocytes) are fragments of the cytoplasm of the megakaryocytes of the bone marrow. They number >140.000/mm3 in the blood of healthy individuals, and are subject to rapid turnover (average lifespan is approx. 7 days). If platelet enter in contact with the collagen or the von Willebrand factor (a protein produced by the endothelium) because of lesions of the endothelial wall, they become activated and acquire the abiility to adhere to the tissue or to each other, thus forming the platelet plug over the lesion(s). Platelets are also activated by thrombin. Activated platelets produce and secrete local hormones called thromboxanes (whose precursor is arachidonic acid), that in turn activate and recruit other platelets, thus amplifying the reaction. Several drugs interfere with platelet activation: e.g. aspirin is an inhibitor of the enzyme cyclooxygenase that starts the pathway converting arachidonic acid to thromboxane.
The process of platelet activation is relevant also to fibrin clotting: this is because activated platelets release coagulation factor V that participates to the activation of thrombin (coagulation factor V is present in the blood in two fractions: a circulating one and one contained in the platelets alpha-granules; it is a protein devoid of protease activity but required for the interaction between prothrombin and factor Xa).
Fibrin clotting. Blood plasma contains 200-400 mg/dL of fibrinogen, a large (MW 340 KDa) soluble protein prduced by the liver. Proteolytic digestion of two short segments (firbinopeptides) of its polypeptide chain at the carboxy- and amino-terminus converts fibrinogen into fibrin. Fibrin is essentially insoluble and readily aggregates forming a network that adheres to the platelet plug. The fibrin network is further stabilized by the formation of covalent isopeptide bonds catalyzed by coagulation factor XIII.
The conversion of fibrinogen into fibrin is due to the specific proteolytic enzyme thrombin. Thrombin is a peculiar protein whose surface glutamic acid residues are post-translationally carboxylated and converted to gamma-carboxy glutamates; it requires calcium to function. The enzyme that gamma-carboxylates glutamic acid uses vitamin K as a cofactor and in avitaminosis K (a rare condition, being this vitamin produced by the intestinal flora) thrombin is produced but is not post-translationally modified and is inactive. Hence deficit of vitamin K (observed during long term antibiotic treatments) or administration of its inhibitors (antivitamin K; e.g. warfarin) cause defective blood clotting.
| TABLE 1: A list of the main coagulation components and the pertinent laboratory tests (due to faulty identifications, not all numbers are used) | ||
Component | Definition and properties | Laboratory test | |
| Components required for the formation of the platelet plug | ||
| Platelets | fragments of the megakaryocyte cytoplasm | platelet count (normal value >150,000 mm3); platelet aggregation test |
| von Willebrand factor | protein present on the vessel wall and in the plasma | vW antigen (measures total vW factor concentration by electroimmunoassay); vW multimer composition |
| Factors required for the formation of the fibrin network | ||
| Factor I | fibrinogen (plasma) | |
| Factor II | prothrombin (plasma) | |
| Factor V | proaccelerin, labile factor (plasma) | specific activity test (measurement of the ability of the patient's plasma to complement the coagulation of a factor V deficient test plasma) |
| Factor VII | proconvertin, stabile factor (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor VII) |
| Factor VIII | antihemophylic factor (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor VIII) |
| Factor IX | Christmas factor, plasma thromboplastin component (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor IX) |
| Factor X | Stuart factor (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor X) |
| Factor XI | Plasma Thromboplastin Antecedent, PTA (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor XI) |
| Flechter factor | prekallicrein (plasma) | |
| Fitzgerald factor | high molecular weight kininogen (plasma) | |
| Factor XII | Hageman factor (plasma) | specific activity test (same as in the case of factor V but test plasma lacks factor XII) |
| Factor XIII | fibrin stabilizing factor (plasma) | measurement of clot stability upon 24 hours incubation in 5 M urea |
| Tissue Thromboplastin | cell surface lipoprotein | |
| Platelet factor 3 | a phospholipid from the platelet membrane | |
| Thrombomodulin | membrane protein of endothelial cells | |
| Intrinsic pathway | fibrinogen, prothrombin, factors V, VIII, IX, X, XI, XIII, prekallikrein, HMW kininogen | Partial Thromboplastin Time (PTT) |
| Extrinsic pathway | fibrinogen, prothrombin, factors V, VII, X | Prothrombin Time (PT) |
| Factors that control coagulation and digest the fibrin network | ||
| Plasminogen | activated by a tissue specific protease to plasmin, digests the fibrin network | Enzymatic activity assay (after activation) |
| alpha2 antiplasmin | ||
| antithrombin III | Specific inhibitor of thrombin | Measured by immunoassay or by its ability to inhibit the enzymatic activity of thrombin |
| Protein C (plasma) | Inhibitor of factors VIIa and Va | Electroimmunoassay |
| Protein S (plasma) | Required for the function of Protein C | Electroimmunoassay |
Hemorragic disorders may be due to three groups of causes, usually quite clearly distinguishable on clinical grounds: (i) defects of the vascular wall (e.g. scurvy, allergic purpura, hereditary hemorragic teleangiectasia, etc.); these are not disorders of the coagulation and all laboratory tests of coagulation are normal. (ii) Platelets disorders, due either to a reduction in number or to functional impairment; the cardinal manifestations of these disorders are prolonged bleeding and purpura, i.e. formation of cutaneous (and internal) petechiae on minor traumas. (iii) Defects of fibrin clotting (most often due to hereditary genetic defects of coagulation factors); the cardinal manifestation is temporary hemostasis (because of the formation of the platelet plug) followed by multiple episodes of re-opening of the lesion and bleeding (because of the dissolution of the platelet plug before any fibrin clot is formed).
Fibrinolysis, the dissolution of the fibrin newtork is an important physiological event, and is due to selective proteolysis. The enzyme responsible for this process is called plasmin and is present in the blood as the inactive precursor plasminogen. Plasminogen is activated by thrombin, thus the coagulation process is auto-regulated.
| Type of abnormality | Pathogenesis | Test(s) for diagnosis |
| Purpuras (insufficient platelet functioning) | Idiopathic Thrombocytopenic Purpura | Low platelet count, in the absence of bone marrow disease (possibly autoimmune cause?) |
| Secondary Thrombocytopenias | Low platelet count, in the presence of bone marrow diseases (e.g. aplasia, leukemia) or accelerated platelet turnover (e.g. allergic, drug induced) | |
| Glanzmann's Thromboasthenia | Normal platelet count, but reduced tendency of the platelets to aggregate (the disease is inherited as an autosomal recessive trait) | |
| von Willebrand disease | Normal platelet count, hereditary deficiency (autosomal, dominant) of the von Willebrand factor, required for the adhesion of platelets to the damaged endothelium | |
| Hemorragic syndromes (impaired coagulation) | Hereditary deficiency of factor VIII (Hemophylia A) | Bleeding and hemorrages; heredity (X-linked, recessive); gene sequencing (the locus of this gene resides in the X chromosome); increased thromboplastin time with normal prothrombin time |
| Hereditary deficiency of factor IX (Hemophylia B) | Bleeding and hemorrages; heredity (X-linked, recessive); gene sequencing (the locus of this gene resides in the X chromosome); increased thromboplastin time with normal prothrombin time | |
| Hereditary deficiency of fibrinogen | Reduced fibrinogen in the plasma associated to mild hemorrages (DIC and hemodilution should be excluded); increased thromboplastin and prothrombin times | |
| Exhaustion due to Disseminated Intravascular Coagulation (DIC) | DIC is an abnormal activation of coagulation due to release of digestive enzymes in the blood (during acute pancreatitis), or to release of the tissue factor of coagulation (e.g. in leukemias and other cancers; in massive traumas), or to coagulase toxins (e.g. snake venom, gram negative sepsis) | |
| Increased coagulation and thrombosis | Disseminated Intravascular Coagulation (DIC) | see above |
| Flebitis | Damage of the vascular wall releases or exposes the tissue factor of coagulation | |
| Impaired circulation in the atria | Blood remaining in the heart atria because of atrial fibrillation or stenosis of the atriventricular valve is at risk of intravascular coagulation because of poor mixing and reduced activation of plasmin | |
| Hereditary deficiency of plasminogen | A rare autosomally inherited disorder associated to DIC and to "ligneous" conjunctivitis (organization of conjunctival microhemorrages) | |
| Hereditary variants of factor V | Some hyperfunctional variants of factor V (e.g. factor V Leiden) may cause hypercoagulation and tendency to thrombosis | |
Laboratory tests of coagulation. The most important test of coagulation is the prothrombin time. Blood is drawn an stored in a tube containing sodium citrate as an anticoagulant (it is important to fill the test tube to the exact volume required since in this test the blood:anticoagulant ratio must be standardized). Citrate prevents coagulation by chelating calcium ions. In the clinical laboratory the plasma is prepared and calcium chloride is added in order to saturate citrate and obtaining a standard concentration of the free calcium ion. Tissue factor is also added and the time required for the precipitation of fibrin is measured by turbidimetry. Typical prothrombin time in healthy adults is in the order of 12-14 sec.
Prothrombin time is increased in the following conditions: administration of warfarin or deficiency of vitamin K; Disseminated Intravascular Coagulation syndrome; liver failure (the liver produces all the coagulation factors); congenital afibrinogenemia; hereditary deficiency of coagulation factors V and X. Prothrombin time is normal in platelet diseases and in hereditary coagulation disorders affecting the intrinsic pathway.
The thromboplastin time is a clinical measurement similar to the prothrombin time, in which the intrinsic pathway is activated by addition of phospholipids and calcium to the patient's plasma. Values in the healthy adult range between 30 and 50 sec. The thromboplastin time is increased in the following conditions administration of warfarin or deficiency of vitamin K; Disseminated Intravascular Coagulation syndrome; liver failure (the liver produces all the coagulation factors); congenital afibrinogenemia; hereditary deficiency of coagulation factors VIII and IX (hemophylias A and B).
Thrombosis is the pathological process of intravascular coagulation. It is usually due to the activation of the extrinsic coagulation pathway in the proximity of a lesion of the vascular endothelium, e.g. over an atheromatous plaque in an artery or in an inflamed vein (thromboflebitis). Arterial thrombi rarely grow to form large structures because of the rapid blood flow; however they may cause infarctions or may become organized (i.e. they may be transformed into scars, rigid because of the deposition of collagen) and cause a permanent stenosis of the affected artery. Venous thrombi are large and fragile structures and may release emboli that may in turn cause arterial obstructions and infarctions.
A very severe condition occurs if the thrombus forms in the left (or more rarely in the right) atrium of the heart, often during chronic atrial fibrillation. A fragment of the thrombus may be expelled in the pulmonary artery and cause massive pulmonary embolism, a condition that is often fatal.
Whenever a risk condition is present, anticoagulant therapy is indicated. Unfortunately risk conditions may not be easy to detect (e.g. flebitis of internal veins).
Home of this course
Slides of this lecture:
 |
 |
 |
 |
 |