Course of LABORATORY MEDICINE
Anemias and hemoglobinopathies
Anemia is a pathological condition characterized by reduced hemoglobin content in the blood. Physiological values of hemoglobin concentration range between >13 g/dL for adult males to >11 g/dL for infants and pregnant women. Hemoglobin concentrations below approx. 9 g/dL are symptomatic, and cause fatigue, dispnea and tachycardia. The increased cardiac work associated to reduced oxigenation of the blood perfusing the myocardium may cause angina pectoris and, occasionally, myocardial infarction. Blood transfusion is indicated when hemoglobin is <7 g/dL.
Anemia is neither a disease, nor a diagnosis: it is a clinical condition that may be present in many different diseases, and may have a number of causes (summarized in the table below). An essential distinction is made between those conditions that impair the biosynthesis of red blood cells and those that reduce the red blood cell lifespan.
| Cause of anemia | Pathogenesis | Test(s) for diagnosis |
| Blood loss: | Acute or chronic hemorrages, internal or external | Demonstration of blood loss (e.g. fecal occult blood; hematuria; hematemesis) |
| Hemolysis, toxic or allergic | Demonstration of hemoglobinuria and reduced erythrocyte lifespan | |
| Deficitary hemopoiesis: | Bone marrow aplasia | Biopsy of the bone marrow |
| Leukemias (and lymphomas) | Demonstration of neoplastic cells in the blood (or the bone marrow) | |
| Talassemias | Demonstration of lack of one of the subunits of hemoglobin in the erythrocytes (by electrophoresis); demonstration of abnormal hemoglobin genes | |
| Chronic kidney diseases | Low erythropoietin levels in the serum; renal failure | |
| Folate or vitamin B12 deficiency | Erythrociytes of increased size (megalocytes); measurement of serum levels of folate; deficit of the intrinsic factor in the gastric secretion | |
| Iron deficiency | Low iron level in the serum | |
| Lead poisoning | High lead concentration in the serum | |
| Reduced erythrocyte lifespan: | Hemoglobinopathies | Abnormal electrophoretic mobility of hemoglobin; demonstration of abnormal hemoglobin genes |
| Talassemias | (see above) | |
| Spherocytosis and ellissocytosis | Direct observation by optical microscopy | |
| Genetic defects of red cell enzymes | Demonstration of abnormal enzyme genes (e.g. Glucose 6-phosphate dehydrogenase in favism) | |
It is important to remark that some diseases cause anemias because of more than a single reason: e.g. thalassemias cause premature death of the red cells but also defective erythropoiesis (hence these diseases appear twice in the table).
The lifespan of erythrocytes does not exceed 120 days, on average: thus every month the bone marrow has to replace one fourth of the circulating red cells. In some diseases causing ineffective erythropoiesis (i.e. destruction of abnormal erythrocytes during their differentiation in the bone marrow) an abnormal expansion of the marrow is observed: e.g. this is the case with beta-talassemia and the so-called "brush" skull. Diseases in which the biosynthesis of DNA is impaired (e.g. deficiency of folate or of vitamin B12) cause a delay in the differentiation of red blood cells, that result fewer, larger and paler (because of lower hemoglobin content) than in the healthy condition.
Anemias caused by reduced erythrocyte lifespan cause a compensatory increase in the rate of red cells production in the marrow, and release of an increased fraction of reticulocytes (immature erythrocytes) in the blood; by contrast reticulocytes are scarce or absent in anemias due to bone marrow aplasia.
As stated above, anemia is not a diagnosis (or it is a very imprecise one), but an easily recognized pathological condition. The identification of the disease underlying a condition of anemia is a differential diagnosis among the conditions listed in table 1, and requires the demonstration of the essential features of the disease. Some relevant cases are individually discussed below.
β - THALASSEMIA (Cooley disease). Thalassemias (or talassemias) are diseases caused by an imbalance of the synthesis of the alpha and beta subunits of hemoglobin. Since hemoglobin is a tetrameric protein, composed by two α and two β subunits, it is necessary that during the maturation of the erythrocyte, the two types of subunit are produced in equal amounts. Thalassemias are genetic diseases due to mutations of the genes of the α or the β subunit of hemoglobin, that may occur in the promoter region of the gene (lowering its transcription efficiency) or in the coding sequence (usually because of the mutation of a coding codon to a stop codon). The aploid human genome contains two copies of the α subunits gene and only one copy of the β subunit gene, on different chromosomes.
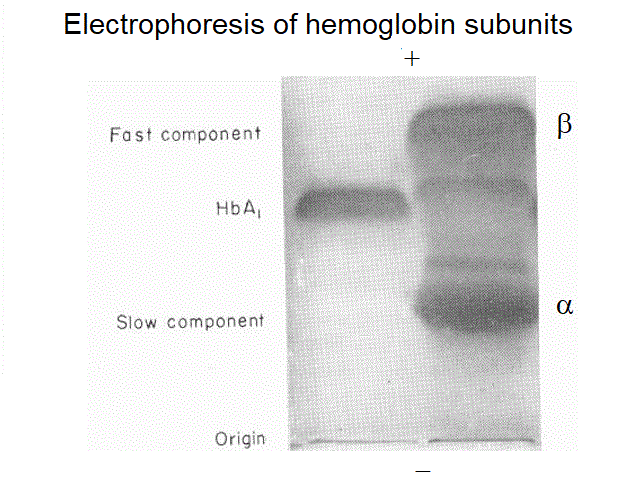
An important indication may be obtained by electrophoresis; this is because the α and β subunits of hemoglobin have different electrophoretic mobility, and in the blood of a thalassemic patient one may observe normal human hemoglobin, and an excess of either subunit, revealed as an additional band with lower (α subunits in β-thalassemia) or higher mobility (β subunits in α-thalassemia).
FOLATE DEFICIENCY. Folate is a vitamin required for the biosynthesis of nucleotides, hence of DNA, and deficiency of folate slows down the proliferation of stem cells of various tissues. The effect is particularly evident for red blood cells because of their rapid turnover, hence the anemia. Due to the reduced rate of replication the erythrocytes are few, large and pale and immature forms (reticulocytes) in peripheral blood are increased. Folate deficiency may occur in pregnant women, due to the high requirement of the vitamin by the foetus, and oral supplementation is indicated.
VITAMIN B12 DEFICIENCY (pernicious anemia, Brill disease)
The biological function of vitamin B12 is somewhat similar to that of folate, since it is a methyl group donor required for the biosynthesis of nitrogen bases of DNA. The pathogenetic mechanism of the anemia and the microscopic appearance of the red cells is also similar, i.e. this is a magalocutic, megaloblastic anemia. At variance with folate deficiency, pernicious anemia also causes neurological symptoms: paraesthesias, irritability, depression, and dementia. The disease is fatal if untreated.
Vitamin B12 deficiency is rarely due to insufficient dietary apport, although this can occur in strict vegan diets. More often vitamin B12 deficiency is due to atrophic gastritis, that impairs the production of the intrinsic factor (a protein required for the absorption of the vitamin) by the gastric mucosa. Because of this peculiarity, which is not shared by any other vitamin, oral administration of vitamin B12 may prove ineffective and intramuscular administration is required.
Diagnosis is suspected on clinical grounds (anemia associated to atrophic gastritis and neurological symptoms) and on the microscopic examination of a blood smear. It is confirmed by the measurement of vitamin B12 concentration in the serum (normal range 20-90 ng/dL). The preferred method for measurement is by radioimmunoassay. Some drugs may interfere with this test: e.g. Colchicine, Neomycin, Para-aminosalicylic acid, and Phenytoin.
IRON DEFICIENCY (sideropenic anemia)
Mammals have no physiological way to eliminate iron, and the iron content in the organism is regulated by its uptake in the gut. Iron is lost with hemorrages and iron deficiency may occur in the presence of insufficient uptake or increased loss by chronic or acute hemorrages. Fertle women have increased iron requirements. The anemia is microcytic (small erythrocytes) and diagnosis is confirmed by low values of sideremia (normal range in male adults: 65-175 ug/dL; female adults 50-170 ug/dL), and low iron saturation of circulating transferrin (normal concentration 200-360 mg/dL; normal saturation > 60%).
REDUCED HEME BIOSYNTHESIS
Insufficient production of the heme is observed in erythropoietic porphyrias and chronic lead poisoning. From a clinical view point these anemias are hypochromic and lack specific chracteristics; however the clinical picture entails several other symptoms (e.g. photosensitivity of the skin in eryhtropoietic porphyrias) that may suggest the diagnosis. Specific laboratory tests are available: measurement of heme precursors in the blood and urine for poprhyrias, measurement of lead in the blood, urine and tissues for lead poisoning.
INSUFFICIENT PRODUCTION OF RED BLOOD CELLS
Insufficient production of red blood cells is caused by diseases that destroy the bone marrow: idiopathic bone marrow aplasia and hematological tumors (e.g. leukemias, lymphomas, etc.). Often these anemias are accompanied by deficiency of other cell types in the blood (reduced platelet count, reduced count of white cells); in leukemias the blood contains tumor-specific cells. The presenting symptom may often be a thrombocytopenic purpura. Diagnosis is suspected because of the results of hemochromocytometric analysis, and is confirmed by bone marrow biopsy.
Hemoglobinopathies are hereditary defects of hemoglobin subunits (the alpha and beta polypeptide chains), usually due to single site mutations. They are inherited as Mendelian recessive traits. Hereditary defects of hemoglobin, due to the high solubility and rich functional behaviour of the protein, are somewhat exceptional with respect to those of other proteins and enzymes. Indeed the most usual effect of random point mutation in proteins is a drastic decrease of stability and solubility and the mutant protein is absent or present at very low concentration in the cell. In hemoglobin this is by no means the only possible effect of mutation and hemoglobinopathies can be classed into at least five groups:
1) Mutations which cause diminished stability. The intracellular Hb precipitates and form Heinz bodies adhering to the red cell membrane. The resulting damage of the membrane reduces the lifespan of the erythrocyte, causing an hereditary anemia.
2) Mutations which cause increased oxygen affinity. The mutant Hb uploads oxygen in the lungs but delivers few oxygen to the tissues, causing chronic ischemia. The ischemic kidney produces more erythropoietin to stimulate the bone marrow and these conditions are often associated to polycytemia (increased blood cell count), increased hematocrit, and increased Hb content.
3) Mutations which cause reduced oxygen affinity. The mutant Hb is not fully saturated with oxygen in the lung capillaries and the blood oxygen content is decreased in spite of a normal arterial oxygen pressure. The patient has cyanosis, but the amount of oxygen released to the tissues is normal and no ischemia is present (in general); the red cell count and hemoglobin content are usually normal or nearly so. The patient has fatigue because oxygen transport under strenuous muscular effort is reduced (a consequence of the reduced arterial oxygen content).
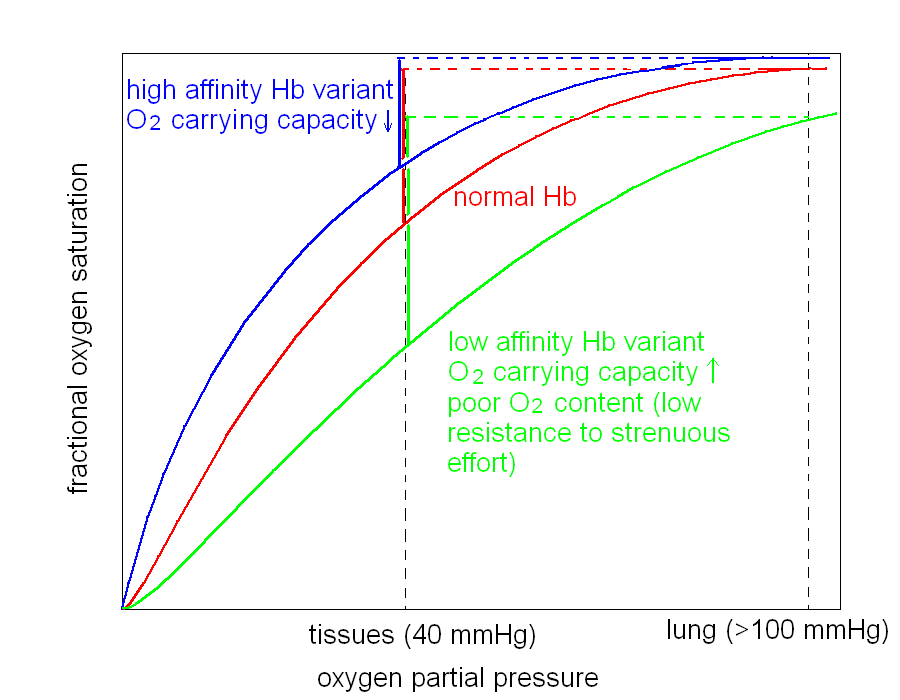
4) Mutations which cause iron oxidation (methemoglobinemias). Mutation of the proximal or distal Histidine of either subunit to Tyrosine causes the heme iron to oxidize to the ferric state. Other mutations that increase the polarity of the heme pocket or allow water to flow in have the same effect. Methemoglobin (Hb with the heme iron in the ferric state) has low oxygen affinity and carries oxygen only on the hemes of the normal subunits, the ferric heme iron being unreactive. Thus the oxygen carrying capacity is halved. The patient presents a characteristic brown cyanosis of the mucosae (black mouth), due to the brownish colour of metHb; other symptoms are as in the case of reduced affinity Hbs, and polycytemia may be present to compensate for the reduced oxygen carrying capacity.
5) Mutations that cause other effects. This is a residual category that includes many different conditions. An important one is sickle cell Hb, the mutation of the sixth residue of the beta chain from Glu to Val. This mutation causes deoxyHb to polymerize and the resulting Hb polymer distorts the red cells, which assume a characteristic sickle shape. Sickled cells may stack in the venulae blocking the circulation: painful microinfarctions follow.
MOLECULAR MECHANISMS OF HEMOGLOBINOPATHIES
Hemoglobin is among the most deeply understood proteins, and hemoglobinopathies have significantly contributed to the study of structure-function relationships in hemoglobin. The macromolecule is a heterotetramer made up of two α and two β subunits arranged in a α2β2 tetramer. The α and β subunits are globular proteins and are made up of 8 (or 7) α-helices. The subunits are arranged in the tetramer in a distorted tetrahedral geometry, and two types of α-β contacts occur. The α1-β1 contact involves helices B, G and H from both participating subunits; the α1-β2 contact involves helix C and the interhelical FG corner from both participating subunits. The regulation of oxygen affinity and cooperativity largely depends on (i) the residues lining the heme pocket of each subunit, (ii) the residues at the α1-β2 interface, which is responsible for the quaternary structural change associated to cooperative oxygen binding, and (iii) the two C-terminal residues of each subunit, that in deoxy Hb pack in proximity of the α1-β2 interface. The above listed regions of the macromolecule are thus those where single aminoacid mutations tend to exhert the most important functional effects. Examples of high oxygen affinity mutants are: Hb McKees Rocks, that bears the deletion of the last two C-terminal residues of the β, and Hb Chesapeake in which α Arg 92(FG3) is replaced by Leu; both mutations destabilize the low affinity T quaternary conformation and favor the transition to the high affinity R conformation. Examples of low oxygen affinity mutants are Hb Presbyterian that bears the mutation β 108 Asn to Lys, in proximity of the FG corner, and Hb Kansas that bears the mutation β 102 Asn to Thr; in the former the affinity is reduced because of stabilization of the T quaternary structure, in the latter presumably because G4 Asn is a heme contacting residue.
Several hemoglobinopathies, together with other hereditary diseases of the red cell (e.g. glucose 6 phosphate dehydrogenase deficiency, thalassemias) cause the red cell to become fragile and reduce its lifespan. While this effect usually results in an anemia, it may protect the heterozygous patient from malaria, because the damaged red cell does not support the full erythrocytic cycle of the parasite leading to an abortive effect. Because of this reason, in regions where malaria is or was endemic, an abnormally high frequency of any of these conditions may be observed (so called "polymorphism due to heterosis", i.e. higher fitness of the heterozygous carrier state that either the healthy or affected homozygous individual).
DIAGNOSIS OF HEMOGLOBINOPATHIES
The diagnosis is suspected on the basis of the clinical information, and confirmed by the finding of a hemoglobin with abnormal electrophoretic mobility. Protein sequencing and finger-printing are nowadays obsolete, and the diagnosis of the specific mutation depends on the sequencing of the genes coding for the α and β subunits.
Home of this course