Course of LABORATORY MEDICINE
Nucleotide Metabolism
Nucleotides play important roles in the cell, both as the monomers of nucleic acids (DNA and RNA) and in their free form (e.g. ATP, cAMP, etc.). They are made up of a pentose sugar (ribose in RNA and ribonucleotides; deoxyribose in DNA and deoxyribonucleotides), an organic heterocyclic base similar to either pyrimidine or purine, and phosphoric acid.

BIOSYNTHESIS OF NUCLEOTIDES
All the nucleotide components can be synthesized ex-novo.
Ribose 5'-phosphate is produced from glucose in the penthose phosphate pathway and is converted to 5'-phosphoribosyl 1'-pyrophosphate (PRPP) by the enzyme ribose phosphate pyrophosphokinase. Deoxyribose is not produced directly; rather ribonucleotides are synthesized and converted to deoxyribonucleotides by the enzyme ribonucleotide reductase that uses the small protein thioredoxin as the reductant. Oxidized thioredoxin is reduced by the NADPH-dependent enzyme thioredoxin reductase, whose inhibition (by drugs and poisons) slows down cell replication.
Pyrimidine bases are produced in a specific biosynthetic pathway from carbamyl phosphate and aspartate; phosphoribosyl pyrophosphate is used as the donor of ribose (the enzymes or metabolytes most commonly affected by heritable diseases are underlined):
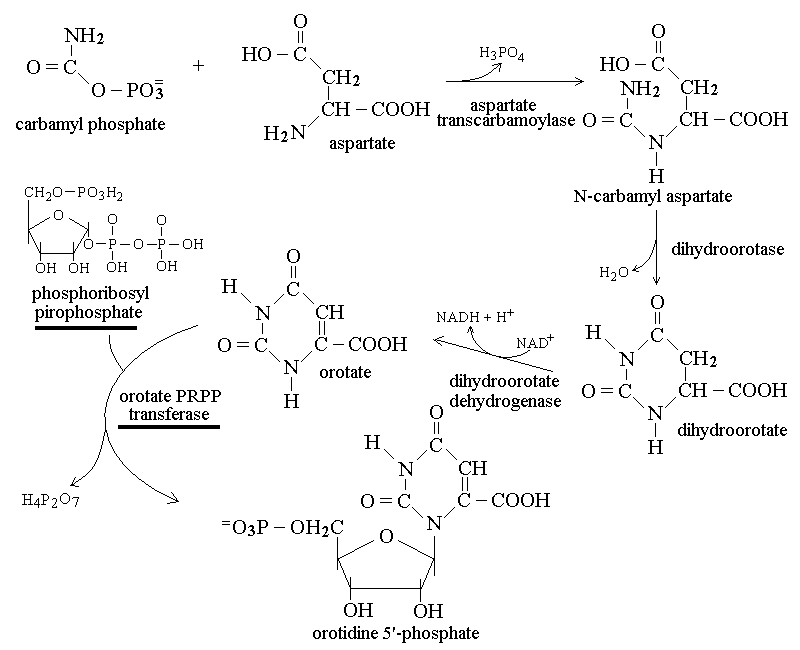
Purine bases are produced in a specific biosynthetic pathway from glycine and aspartate; phosphoribosyl pyrophosphate is used as the donor of ribose:

SALVAGE PATHWAYS
Given that the biosynthesis of nucleotide bases is energy-expensive, all animals (man included) possess biochemical pathawys that allow the recovery of nucleotides derived from DNA degradation (either because of dietary apport or because of cell and tissue turnover). In man two enzymes are responsible for the salvage of purine bases: adenine phosphoribosyl transferase and hypoxantine-guanine phosphoribosyl transferase. The chemical reactions catalyzed by these anzymes are as follows:
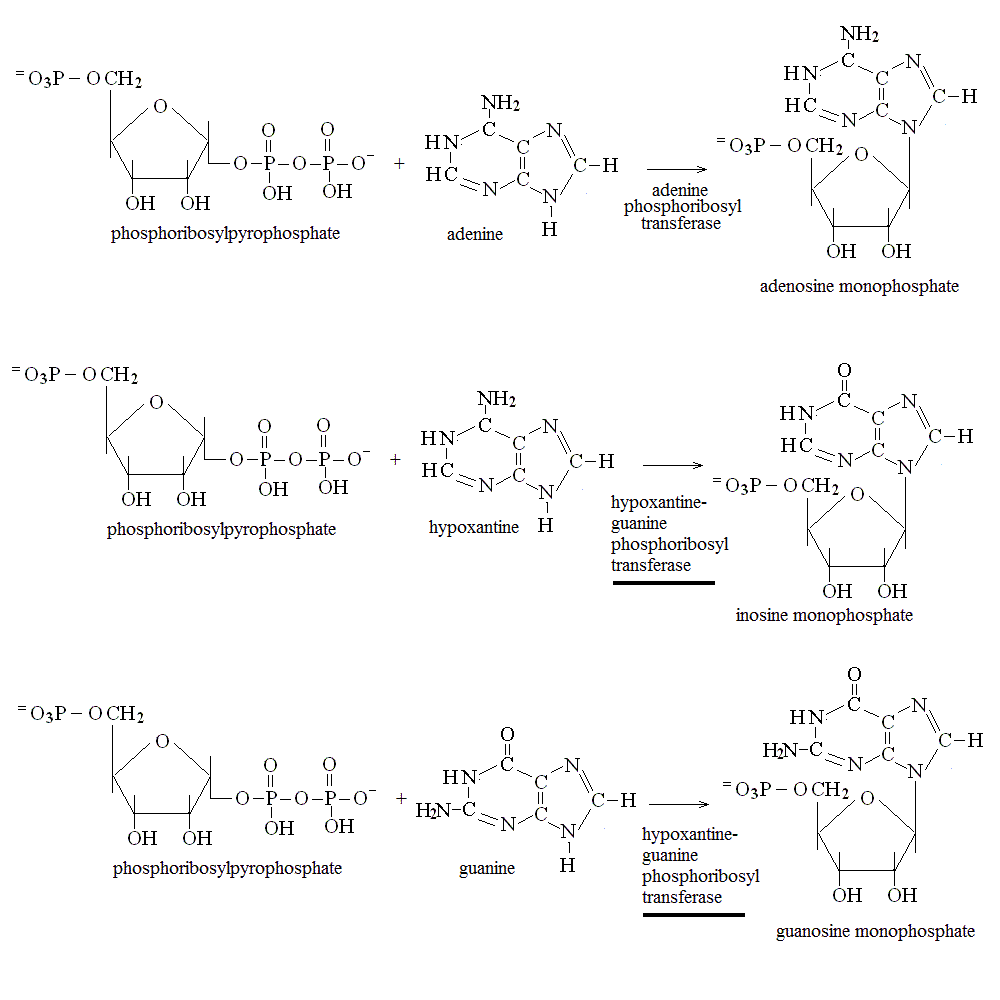
DISTURBANCES OF THE BIOSYNTHESIS AND SALVAGE OF NUCLEOTIDES
All the metabolic pathways involved in the biosynthesis of nucleotides can be subject to hereditary disturbances; however most defects in such important bisynthetic pathways are likely to be non viable and thus few diseases are described.. The most common inherited disturbance of the pentose phosphate pathway, responsible for the biosynthesis of ribose, is the genetic anomaly of glucose-6-phosphate dehydrogenase known as favism, which is described under the diseases of glucose metabolism.
The most important disease of nucleotide biosynthesis/salvage is the hereditary, X-linked deficiency of hypoxantine-guanine phosphoribosyl transferase. The genetic defect varies and, depending on the amount of the enzyme that is produced and its activity, the severity of the resulting disease also varies from mild sex-linked uric aciduria (a variant of gout, see below), to the lethal Lesch Nyhan syndrome, associated with renal insufficiency, severe mental retardation, automutilation and death in the infancy. Diagnosis is based on clinical grounds and is confirmed by genetic analysis.
Another important hereditary disease of nucleotide bisynthesis is Orotic aciduria, that causes megaloblastic anemia, mental retardation, and stunted growth. This condition is due to hereditary deficiency of the enzyme orotate phosphoribosyl transferase, a bifunctional enzyme that catalyzes two reactions in the pyrimidine biosynthesis pathway: conjugation of orotic acid to ribose 5'-phosphate and decarboxylation of orotidine 5-phosphate to uridine 5-phosphate. The disease may be suspected because of the finding of a megaloblastic anemia in the newborn, resistent to the administration of folate and vitamin B12, and is confirmed by the finding of high amounts of orotic acid in the urine. Gene sequencing is also possible and reveals the mutation.
Increased activity of Phosphoribosyl Pirophosphate Synthetase is a rare hereditary, X-linked genetic defect causing a genetic form of gout. The enzyme is allosteric and downregulated by ADP and 2,3-DPG. The abnormal variant is constitutively active, irrespective of the concentration of its allosteric inhibitors. The disease may be apparent also in female newuborns, given that the effect of the mutation is dominant. Diagnosis is by gene sequencing.
Adenylosuccinase Deficiency is a rare hereditary defect, associated with mental retardation. Diagnosis is established by gene sequencing.
Differential diagnosis
Disturbances of the biosynthesis and salvage pathways cause diseases in which the following symptoms predominate: mental retardation, gout, megaloblastic anemia. The symptoms appear very early, within the first months of life. Aside from the case of Lesch Nyan syndrome, in which major neurological and psychiatric symptoms predominate, the other conditions are quite uncharacteristic. The main difficulty for diagnosis is that the physician rarely considers these hereditary defects as possible causes of stunted growth and mild mental retardation.
In a newborn or infant presenting stunted growth, a blood test should be required as a routine analysis. Finding of megaloblastic anemia is an important indication, because this condition occurs in folate or B12 deficiency or in hereditary diseases causing slowed cell cycle (reduced rate of DNA biosynthesis). Indeed the molecular mechanism underlying megaloblastic anemia is the same for the two conditions, because folic acid and vitamin B12 are required cofactors for the biosyntehsis of nucleotides. Folate deficiency in the newborn occurs under conditions of severe social deprivation and usually is associated to malnutrition of the infant and his/her parents. Vitamin B12 deficiency in the newborn or infant may be due to malnutrition, or to ideological factors: e.g. vegan parents may undergo mild vitamin B12 deficiency, which, through lactation may affect their baby. However, the serum concentration of both folate and vitamin B12 can be determined: if these are low, the diagnosis is of a deficiency megaloblastic anemia, if they are normal, a hereditary condition should be suspected and investigated by gene sequencing.
| disease | inheritance | biochemical defect | symptoms | laboratory findings |
| Lesch Nyan s. | X-linked (male only) | hypoxantine-guanine phosphoribosyl transferase (purine salvage pathway) | gout, mental retardation, psychotic autoaggressive symptoms | increased urate serum |
| orotic aciduria | autosomal recessive | orotate phosphoribosyl transferase (pyrimidine biosynthesis pathway) | megaloblastic anemia, mental retardation, stunted growth | orotic acid in serum and urine |
| Increased activity of Phosphoribosyl Pirophosphate Synthetase | X-linked, dominant (female affected) | Phosphoribosyl Pirophosphate Synthetase (purine and pyrimidine biosynthesis pathway) | familial gout | increased urate serum |
| Adenylosuccinase Deficiency | autosomal recessive | Adenylosuccinate lyase (purine biosynthesis pathway) | mental retardation, epilepsy | succinylated purine derivatives in the urine |
| Severe Combined Immunodeficiency Disease (SCID) | autosomal recessive | Adenosine Deaminase (ADA; purine degradation pathway) | repeated infections (may be lethal in the absence of bone marrow transplant) | reduced white cell counts; greatly reduced immunoglobulin fraction in the serum electropherogram; reduced ADA activity in the hemolysate |
| Purine Nucleoside Phosphorylase Deficiency | autosomal recessive | Purine Nucleoside Phosphorylase (purine degradation pathway) | immunodeficiency; repeated infections | reduced T lyphocyte count |
| Pyrimidine 5' Nucleotidase Deficiency | autosomal recessive | Pyrimidine 5' Nucleotidase (pyrimidine degradation pathway) | hemolytic anemia | basophilic stippling in the erytrocytes due to accumulation of pyrimidine nucleotides |
| Dihydropyrimidine Dehydrogenase Deficiency | autosomal recessive | Dihydropyrimidine Dehydrogenase (pyrimidine degradation pathway) | mental retardation | elevated pirimidine bases in the serum |
NUCLEOTIDE CATABOLISM
Excess nucleotides introduced with the diet or produced by tissue turnover are degraded in specific biochemical pathways. The terminal metabolytes of pyrimidine bases are derivatives of 3-amino propanoic acid:
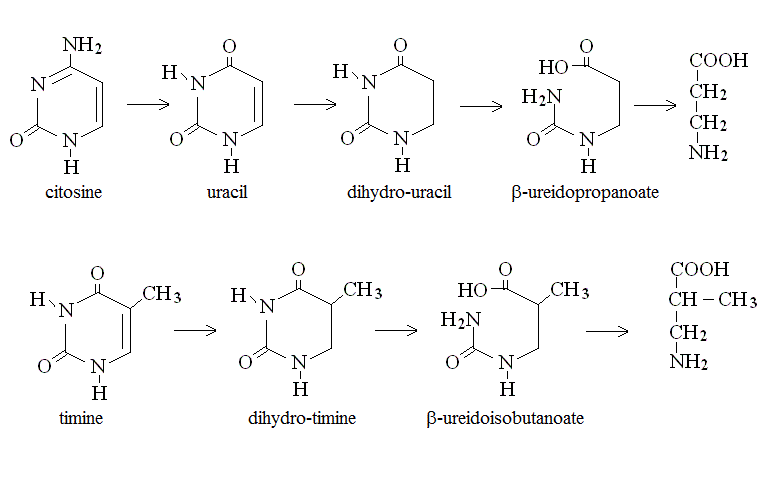
Purines are converted to uric acid which is excreted by the kidney:
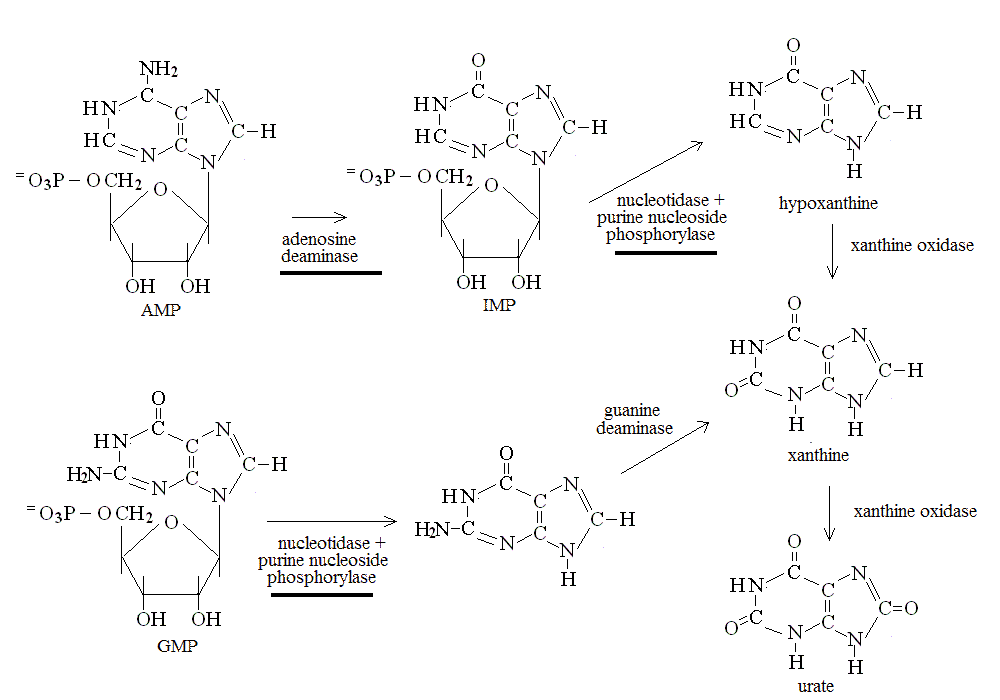
GOUT
Gout is a relatively common metabolic syndrome caused by increased serum concentration of uric acid, the terminal metabolyte of purine degradation (see above). There are different possible causes of hyperuricemia and gout, which therefore cannot be considered a precisely identified disease; rather it is a syndrome that may appear because of: (i) hydiopathic hyperuricemia (hydiopatic, meaning "of no known cause" in this case is probably to be explained as the effect of an unfavorable combination of allelic variants of otherwise functional enzymes in the purine degradation pathway); (ii) increased biosynthesis of purine bases (e.g. myeloproliferative diseases like leukemias and lymphomas; psoriasis); (iii) reduced renal clearance of uric acid (e.g. renal insufficiency); (iv) metabolic conditions such as reduced purine salvage (e.g. hereditary deficiency of hypoxantine-guanine phosphoribosyl transferase). Gout may be aggravated by a diet rich in nucleic acids (e.g. fish, meat) or alcohol and by obesity and diabetes.
Considerations on hydiopathic hyperuricemia: it has been repeatedly asserted that "hydiopathic" is an elegant word to conceal our ignorance and that its meaning is "we do not know why". Nowadays, however we have quite a clear idea of what hydiopathic means, or could mean, at least in clinical contexts like hyperuricemia. The members of the human population differ because of the presence in their genomes of different allelic variants of the same genes. Even when these variants are fully functional and none can be considered pathological, there will be people having more functional combinations of alleles of different genes and people having less functional combinations. Thus the human population includes individuals who produce and excrete uric acid at a faster or a slower rate, and the urate concentration in the blood serum is distributed as a gaussian. This, coupled with the different dietary intake of purine bases, explains the hydiopathic hyperuricemia as the tail of the gaussian distirbution. A quantitative example of such reasoning is presented elsewhere.
Clinical features: gout usually manifests itself with a sudden and painful arthritis of one or more major joints (ankle, knee, elbow, wrist); involvement of the metatarsophalangeal joint of the first toe is common (podagra). Precipitating factors may be minor traumas, or administration of some drugs (penicillin, diuretics). The affected joint is actuely painful, red and swollen and on palpation characteristic hard calculi (gout tophi, consituted by urate salts) may be appreciated on palpation. Fever is common. Chronic cases present erosive joint deformities.
Diagnosis: the clinical presentation is quite typical and suggest the correct diagnosis, which is confirmed by the finding of increased serum urate concentration (normal value in adult males < 7 mg/dL; this value is very close to the solubility of urate in the plasma), and of negatively birefringent needle-shaped urate crystals in the synovial fluid (birefringence is the property of a crystal whose refractive index varies with the direction of light polarization: observed in the microscope upon polarized light the crystal changes from bright to dark as the polarizing filter is rotated). Urate concentration is measured using an enzymatic assay (the enzyme uricase is used, that converts uric acid to allantoin; the reaction is monitored by absorbance spectroscopy at 293 nm; uricase is not produced by humans). Once a diagnosis of gout has been made, the possible underlying conditions should be looked for (e.g. leukemia, diabetes, etc.).
DIFFERENTIAL DIAGNOSIS OF GOUT: gout is a non-febrile recurring acute arthritis, to be differentiated from septic arthritis, rheumatoid arthritis, rheumatic fever, autoimmune diseases. Increased urate in the serum and the urine and the presence of tophi are diagnostic. Moreover in gout autoantibodies characteristic of rheumatoid arthritis and other autoimmune diseases are absent. Rheumatic fever follows a streptococcus infection and the patient has elevated indexes of it (e.g. high Anti-Streptolysin O titer, ASO). Acute septic arthritis is a major infectious syndrome with fever and leukocytosis.
Therapy of the acute attack is very specific, as gout responds dramatically to colchicine (not more than 3-4 mg/die).
DISTURBANCES OF THE DEGRADATION OF NUCLEOTIDES
Several hereditary diseases due to defects of nucleotide degradation are known.
Severe Combined Immunodeficiency Disease (SCID) is the common name of several hereditary deficits. The most common form is a X-linked defect of the of IL2RG gene that codes for the so-called common gamma chain, a component of several receptors of the lymphocyte membrane. This form of the disease is not related to nucleotide metabolism. An autosomal recessive form of SCID is due to the genetic defect of adenosine deaminase, the enzyme that converts AMP to IMP and starts the degradation of adenosine. Pathogenesis is due to the accumulation in the cells of deoxyAdenosine triphosphate (dATP), that inhibits the enzyme ribonucleotide reductase and slows down the biosyntehsis of DNA. All rapidly regenerating tissues are affected and the precursors of granulocytes and lymphocytes are affected most. The disease is invariably fatal because of infections; therapy usually requires bone marrow transplant. Diagnosis is suspected on clinical grounds (severely reduced lymphocyte and granulocyte count) and confirmed by genetic analysis. Notice that since several forms of SCID exist, which cannot be distinguished on clinical grounds, the differential diagnosis relies on the type of inheritance, the demonstration of the biochemical alteration, and gene sequencing.
Purine Nucleoside Phosphorylase Deficiency. Purine Nucleoside Phosphorylase is responsible for releasing inosine or guanosine from the corresponding ribonucleotides or deoxyribonucleotides at the very beginning of the purine degradation pathway. The disease causes accumulation of deoxyribonucleotides, especially dGTP and affects mostly the replication of T-lymphocytes. The clinical picture is that of an immune deficiency, less severe than SCID.
Pyrimidine 5' Nucleotidase Deficiency is an hereditary disease whose main symptom is an hemolytic anemia of variable severity.
Dihydropyrimidine Dehydrogenase Deficiency causes neurological symptoms and mental retardation. Diagnosis is based on the finding of elevated concentration of free pyrimidine bases in the serum.
DIFFERENTIAL DIAGNOSIS OF GENETIC DISTURBANCES OF PURINE AND PIRIMIDINE METABOLISM
Genetic disturbances of the biosynthesis and degradation of nucleotide bases may cause at least three widely different clinical syndromes:
(i) mental retardation, possibly associated to gout and/or stunted growth. The Lesch Nyan syndrome is the severest form of this group.
(ii) Congenital immunodeficiency syndromes. SCID due to ADA deficiency is the severest and paradigmatic example.
(iii) Anemias, either megaloblastic or hemolytic.
The differential diagnosis of mental retardation is difficult because this condition may be due to a host of different causes, not all of them known.
A large number of congenital immunodeficiencies is described. Some of these affect selectively the B or T lymphocytes, others affect both. The crucial step is differentiating from acquired immunodeficiencies (e.g. congenital HIV/AIDS). The immunodeficiency usually becomes clinically evident with weaning, because the mother's milk contains immunoglobulins that the breast-feed newborn absorbs. It is uncommon for acquired immunodeficiencies to appear as early as during weaning, thus an hereditary cause should be considered for every early-onset immunodeficiency, making genetic testing mandatory.
Slides of this lecture:
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
Home of this course