| Course of LABORATORY MEDICINE Disturbances of Aminoacid Metabolism Proteins ingested with the diet are digested to aminoacids by endopeptidases (e.g. pepsin, trypsin and chymotrypsin) and by esopeptidases (carboxypeptidases and aminopeptidases) and absorbed by the small intestine. They constitute the major source of nitrogen in our metabolism. Endogenous proteins are degraded to aminoacids by several systems (e.g. the proteasome, and the lysosomes); these may be used to produce new proteins or can be directed to the Krebs cycle to produce energy. Since the Krebs cycle does not consume nitrogen, the catabolism of aminoacids requires the removal of the amino group. This is achieved by enzymes called transaminases (or aminotransferases). There are as many transaminases as there are aminoacids, and each uses alpha ketoglutaric acid (a metabolyte of the Krebs cycle) as the acceptor of the amino group; the reaction of Alanine Aminotransferase (ALT; also called Glutamic Piruvic Transaminase, GPT) is as follows: 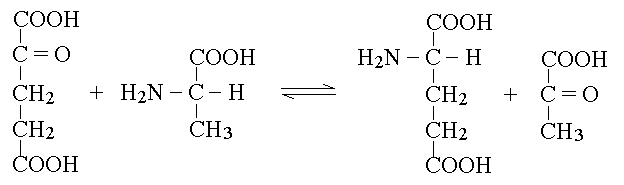 The reaction product is piruvate, which proceeds to the Krebs cycle after decarboxylation by private decarboxylase. The reactions catalyzed by transaminases tend to accumulate glutamic acid and to subtract alpha-ketoglutarate from the Krebs cycle. Recovery of alpha-ketoglutarate is achieved by oxidative deamination. The reaction, catalyzed by glutamate dehydrogenase, releases free ammonia: 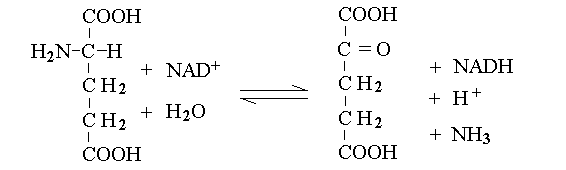 Alpha-aminoacids are thus converted to the corresponding alpha-ketoacids, and their amino group is released as ammonia. Some animals (notably fishes) eliminate ammonia in the urine: they are defined ammoniotelic (or ammonotelic). Unfortunately, ammonia is basic and at high concentration it is toxic for the renal tubules: thus animals not living in water must convert this compound to something less toxic in order to be able to concentrate it in the urine a to save water. Mammals are defined ureotelic because they use urea as the terminal metabolyte of nitrogen, and their blood and urine contain this compound as the main source of non-protein nitrogen. The transformation of ammonia into urea is achieved by a metabolyc pathway called the urea cycle: 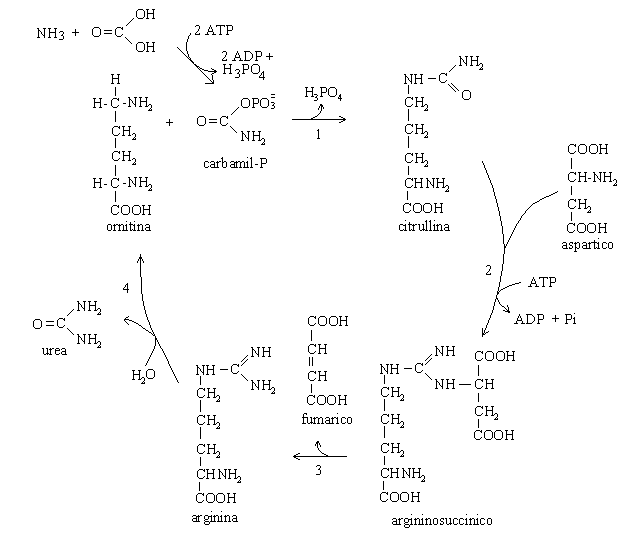 The first reaction (catalyzed by the ezyme carbamoyl phosphate synthetase) uses bicarbonate, ammonia and 2 molecules of ATP to produce the high energy intermediate carbamoyl phosphate. Carbamoyl phosphate is then combined with a molecule of the non-proteic aminoacid ornithine to produce the non-proteic aminoacid citrulline; the enzyme is called Ornithine transcarbamoylase. Citrulline is combined with a molecule of aspartic acid by the enzyme Arginino-succnate synthetase; the product is argininosuccinic acid. The enzyme Arginino-succinate lyase breaks down this molecule to yield arginine (a proteic aminoacid) and fumarate (a Krebs cycle metabolyte). Arginase, the last enzyme of the cycle hydrolyzes a C-N bond of arginine to produce urea and ornithine, than can be used in a new cycle. The overall balance of the urea cycle (neglecting the consumption of 3 ATP) is as follows: 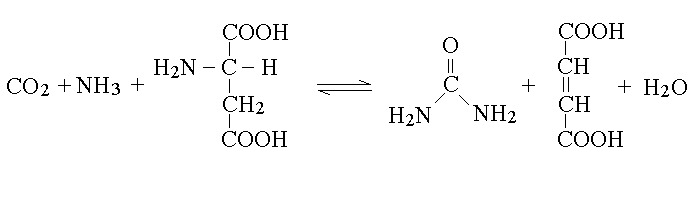 An interesting observation is that only one of the nitrogens of urea comes from ammonia (via glutamate dehydrogenase); the other derives from one molecule of aspartic acid. Clearly aspartic acid is not enough to sustain the urea cycle: it accounts for only some 7% of the total aminoacids of alimentary proteins. Where does aspartic acid come from? The amino groups of all aminoacids are transferred to alpha-ketoglutarate by the corresponding aminotransferase, and aspartate is no exception. However the transaminase reactions are all reversible and in the case of Aspartate Aminotransferase (AST; also called Glutamate Oxaloacetate Transaminase, GOT) the reaction flows mainly backwards: 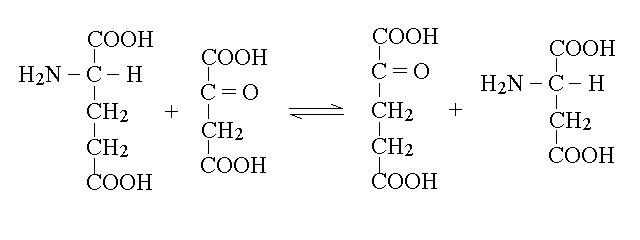 As a consequence, aspartate is produced from glutamate and oxaloacetate (a Krebs cycle metabolic intermediate) and the two nitrogens of the molecule of urea derive from all aminoacids that our organism catabolyzes, via transamination to glutamate and aspartate, as schematically depicted in the figure below that shows the flow of nitrogen from aminoacids to urea (in red the key metabolytes). 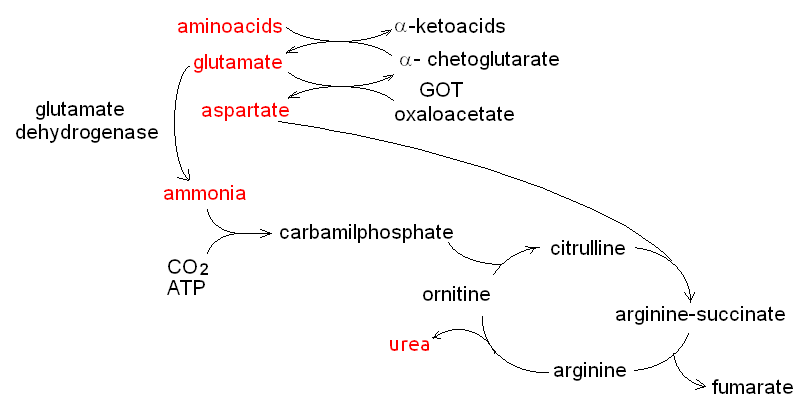 The alpha ketoacids obtained from transamination are further metabolized and are converted to acetyl-CoA or to intermediates of the glycolysis or the Krebs cycle. The relationships between these catabolic pathways is fascinating (but quite intricated): 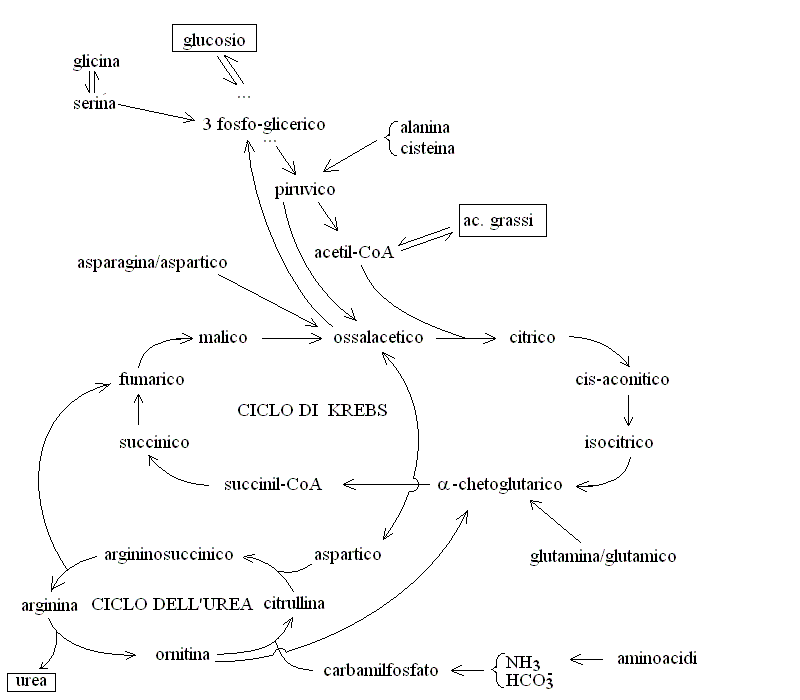 Nitrogen is contained in the blood in two major forms: urea and proteins. Less relevant sources are free ammonia, free aminoacids, hormones and creatinine. The normal range for BUN is 6-20 mg/dL. It is important to remark that, given the chemical formula of urea (H4CN2O, MW=60) and its nitrogen content (47% w/w), 6-20 mg/dL of BUN correspond to 13-43 mg/dL urea. The easiest method to determine BUN is as follows: (i) serum is obtained from venous puncture; it may be de-proteinized using TCA precipitation followed by neutralization. (ii) The concentration of free ammonia is selectively measured using the appropriate electrode. (iii) The bacterial enzime urease is added to the sample; it catalyzes the decomposition of urea to carbon dioxide and ammonia: CO(NH2)2 + H2O --> CO2 + 2 N3. (iv) The concentration of ammonia is again determined with the same electrode; BUN corresponds to the increase of ammonia concentration after addition of urease. In alternative several colorimetric methods are available, based on the Bethelot's reaction: urea is degraded to carbon dioxide and ammonia by urease and ammonia is allowed to react with hypochlorite and phenol (in the presence of a catalyst) to yield the blue compound indophenol, whose concentration can be determined by absorbance spectrophotometry. Creatine and creatinine. Creatine is synthesized by the liver starting from arginine, glycine and S-adenosilmethinine. The daily production of creatine is approximately 1 g/die. This compound is used mainly by the skeletal muscle as a reservoir of high energy phosphate bond, in the form of phosphocreatine (or creatine phosphate), that can rapidly and anaerobically replenish ATP (for burst energy consumption). Phosphocreatine may undergo spontaneous conversion to the terminal metabolyte creatinine, which cannot be utilized by the muscle and is secreted in the blood and eliminated in the urine. The formulas and reactions are as follows: 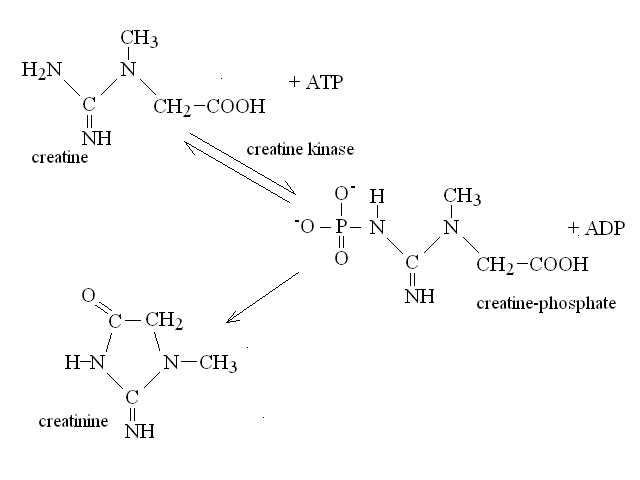 Creatinine is the second most important low-molecular weight nitrogen containing compound in the blood. In the healthy adult its serum concentration is 0.5-1.2 mg/dL (or 50-110 uMol/L). Measurement of renal function. Since urea and creatinine are cleared by the kidney, an increase of the serum concentration of these substances is usually indicative of kideny disfunction. Azotemia is essentially a synonimous of BUN, and hyperazotemia is the clinical condition in which azotemia is increased above normal values. Creatinine clearance. The kidney's ability to excrete creatinine is easily measured and provides an important estimate of the organ's function. The method takes advantage of the almost constant concentration of creatinine in the serum, due to the constant rates of production and excretion. A sample of urine is collected over a precisely measured time (e.g. one hour), preferably by a catheter. The concentration of creatinine is measured in the serum and in the urine. Given that the volume of urine can be precisely determined, one can calculate the moles or milligrams of creatinine excreted in one minute by the kidneys: moles of creatinine excreted / minute = [urinary creatinine] x volume of urine / time The amount of creatinine thus calculated was contained in the volume of serum filtered by the kidneys in one minute, i.e.: volume of serum filtered / minute = moles of creatinine excreted in one minute / [serum creatinine] The normal clearance of creatinine is 120 mL / minute; since creatinine is neither reabsorbed nor excreted by the renal tubuli, this value equals the volume of serum filtered by the glomeruli in one minute. Renal atrophy, polycystic kidney disease, glomerulonephritis are common causes of reduced glomerular filtration. It is important to remark that the volume of urine produced in one minute is highly variable and does not provide any information on glomerular filtration: the kidney can reabsorb up to 99% of the filtered water, depending on the amount of water the patient drinks and the osmotic effect of urine solutes. It is perhaps surprising that a metabolism so fundamental as the Urea cycle and aminoacid catabolism is so prone to genetic defects, given that these diseases usually cause early death and are strongly counter selected. However one should consider that (i) insufficiency of dissimilatory pathways (such as the urea cycle) is usually silent in the foetus, as the mother body takes care of the excretion of the accumulated metabolyte; and (ii) these diseases are recessive and in some cases X-linked; thus the affected genes are to some extent protected from severe selection.
Home of this course |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||