| Course of LABORATORY MEDICINE Disturbances of Glucose Metabolism Glucose is among the most fundamental nutrients for humans. It is present as such in some fruits (e.g. grapes), and as a monomer of di-and poly-saccharides in many foods (e.g. saccharose in fruits, lactose in milk, starch in cereals). As di- and poly-saccharides are not absorbed by the intestine, digestion (i.e. hydrolytic depolymerization) is necessary. Starch is digested by amylases from the pancreas and the salivary glands; disaccharides are digested by the four disaccharidases on the membrane of intestinal cells (maltase, isomaltase, saccharase, and lactase). Monosaccharides are adsorbed by the intestine and released in the superior mesenteric vein. The liver removes them from the blood, converts them to glucose if necessary, and stores glucose as glycogen (approximately 50g overall, corresponding to the daily requirement of the brain). All organs utilize some glucose for energy production and some of them use only glucose (e.g. the brain). It is of critical importance that the blood concentration of glucose is maintained constant. Glycemia, the blood glucose concentration, in the healthy fasting adult ranges between 70 and 100 mg/dL. Glycemia is regulated by the combined action of several hormones, among which insulin (a protein produced by the Langerhans islets in the pancreas) is the most relevant. Insulin causes the tissues to absorb glucose and to utilize it for their metabolism, and the liver to release glucose from glycogen; excess insulin causes hypoglycemia (lower tha normal glucose concentration in the blood), lack of insulin causes hyperglycemia (excess glucose in the blood). Other hormones that contribute to the control of glycemia are glucagone (another protein produced by the Langerhans islets) and cortisol. Glucose is metabolyzed via two main energy producing pathways, namely glycolysis (followed by the Krebs cycle) and the penthose phosphate pathway. The latter fulfills the additional function of producing ribose-5-phosphate which is necessary for the biosynthesis of nucleotides. Both pathways involve redox reactions and reduce NAD+ (or NADP+) that require oxygen to be reoxidized; however glycolysis can be carried out to some extent in excess of the availability of oxygen (anaerobic glycolysis). If this happens (e.g. during strenuous muscular effort), NAD+ is regenerated by the enzyme lactate dehydrogenase which reduces piruvate, the end product of glycolysis, to lactate. Lactate is then released in the blood and taken up by the liver which converts it back to glucose by glyconeogenesis (Cori's cycle). Except for a couple of reactions, glyconeogenesis is the reverse of gycolysis and uses the same enzymes; it is however a energy-intensive process, whose cost is approximately one fourth of the total lactate available. Glyconeogenesis requires piruvate or other glycolysis intermediates, that can be obtained by sources other than lactate: e.g. alanine (which is converted to piruvate by Alanine Aminotransferase); aspartate (which is converted to oxaloacetate by Aspartate Aminotransferase and then to phosphoglycerate); serine (which can be converted to phosphoglycerate); etc. Thus, insufficient dietary apport of glucose can be counteracted by degradation of endogenous proteins and conversion of some of their constituent aminoacids to intermediates of the glyconeogenesis. By contrast, humans (and other animals) have no metabolic pathway that converts fatty acids or their metabolytes to glucose (with the possible exception of the metabolism of propanoyl-CoA). This conversion is not impossble though: some plant seeds possess the necessary enzymes (the so-called glyoxalate pathway); it has been lost during evolution because animals obtain excess sugars from their diet and there is no selective advantage in being able to synthesize them form fat. The measurement of glucose concentration can be effected with several techniques; the preferred one takes advantage of the specificity offered by enzymes. Chemical methods are economically very convenient usually but are at a loss in discriminating glucose from its epimers (e.g. galactose). An example of a typical enzymatic method is as follows: 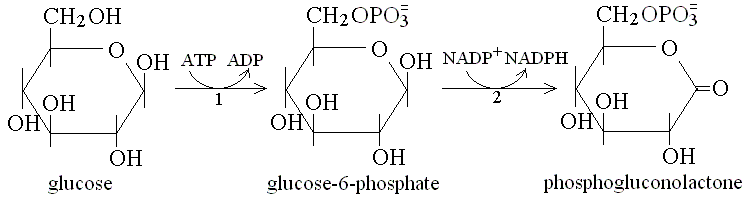  A specific test for diabetes is the Glucose Tolerance Test. To perform this test the glycemia is measured in the fasting patient, then glucose is administered orally at a dose of 1.75 g/kg as a 25 g/dL aqueous solution, and glyecemia is measured again at 30 min intevals for at least 2 hours. Typical glycemia profiles for healthy adults and Insulin Dependent Diabetes Mellitus (IDDM) patients are reported in the figure below (attention: significance interindividual variability!): 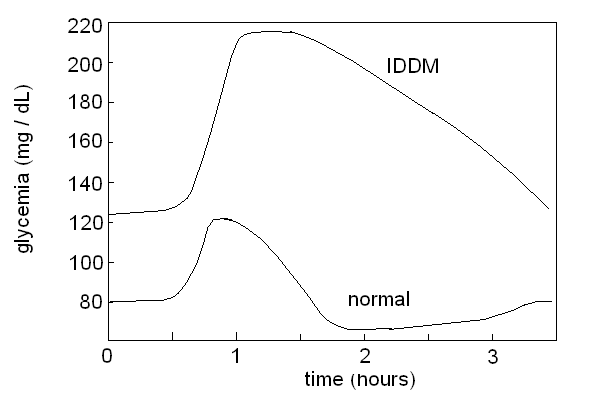 The glycemic index is a measure of the ability of glucose containing foods to cause a rapid increase of the glycemia. To measure the glycemic index of a given food, the fasting experimental subjects are feed with the amount of food necessary to provide 50 g of sugars and their glycemia is measured at 15-30 min. intervals for two hours. The glycemic index is expressed as the ratio between incremental area of the glycemic curve divided by the area of the control food (50 g of pure glucose), and expressed as a percentage. A high GI (>70%) food is one whose sugars are rapidly absorbed and produces a fast increase of the glycemia and an equally fast decrease, coupled to a strong insulin response; a low GI (=50% or less) food produces a slower and less marked increase of the glycemia and a more gradual insulin response. As a general rule low GI foods are preferable, and high GI foods are to be reduced for diabetic patients.
Autoimmune destruction of the insulin producing cells (B-cells of the Langerhans islets in the pancreas) results in type I diabetes mellitus. The term diabetes (from the greek flow-through) indicates a clinical condition characterized by polyuria (increased urine production) and polydipsia (thirst, increased water uptake); there are several diseases associated to these symptoms. A basic dichotomy is whether the urine contains glucose (diabetes mellitus: in this case diuresis is increased because of the osmotic effect of glucose) or not (diabetes insipidus; diuresis is increased because of reduced reabsorption of water, usually due to insufficient production of the antidiuretic hormone vasopressin by the neurohypophysis). Type I diabetes mellitus accounts for approx. 15% of all cases of diabetes mellitus, but is the most severe form and if untreated is lethal within months or at most a few years after diagnosis. It usually occurs in infants or adolescents, and since it can be effectively treated by subcutaneous administration of insulin at regular time intervals, correct diagnosis is of utmost importance. Strictly speaking, what we call type I diabetes mellitus is not truly a disease: rather it is the anatomophysiological condition resulting from one (the autoimmune destruction of insulin producing cells), in some sense similar to a palsy or to the traumatic loss of a limb. However, since it causes symptoms and it damages several organs, thus exhibiting a progression, it behaves and must be treated like a disease. Diagnostic criteria for IDDM are fasting glycemia >140 mg/dL and peak glycemia in the oral glucose tolerance test >200 mg/dL. However a fasting glycemia >115 mg/dL in the adult or >130 mg/dL in the child strongly suggest further analysis for IDDM and other types of hyperglycemias. The insulin concentration in the serum can be measured (by Insulin RadioImmunoassay, IRI) to demonstrate its decreased or absent production, but this is rarely necessary. Biochemistry and pathophysiology of IDDM (Insulin-Dependent Diabetes Mellitus) are surprisingly complex, and some details remain unclear. The main consequence of the lack of insulin is that most tissues of our body cannot uptake glucose (the brain does not reuire insulin for this and is not damaged), which accumulates in the blood (causing hyperglycemia) and is filtered by the kidney at a rate incompatible with reabsorption (causing glycosuria and osmotic polyuria). Loss of water in the urine causes thirst. Free glucose is chemically reactive, because of its aldehydic group and may combine with the amino groups of lisyl residues of proteins, causing protein glycosylation. Hyperglycemia, though important, is not the cause of the most relevant damages due to IDDM: in fact, the most damaging events occur inside the cells, and are due to lack of glucose, and consequent shift to consumption of fatty acids. The beta oxidation of fatty acids yields acetyl-CoA to fuel the Krebs cycle, thus it can sustain the cell's energy requirements. However the Krebs cycle requires a small but continuous apport of oxaloacetate to replace losses due to removal of metabolytes for anabolic requirements (e.g. alpha-chetoglutarate may be used to produce glutamate and glutamine; oxaloacetate may be used to produce aspartate and asparagine; succinyl-CoA is used to produce delta-aminolevulinc acid, required by heme biosynthesis; etc.). Oxaloacetate can be produced from piruvate by the enzyme piruvate carboxylase, but cannot be produced starting from any metabolyte of the beta-oxidation, with the exception of the small amount of succinyl-CoA produced during the metabolysm of propanoyl-CoA (in the case of the rare fatty acids with uneven number of carbons). 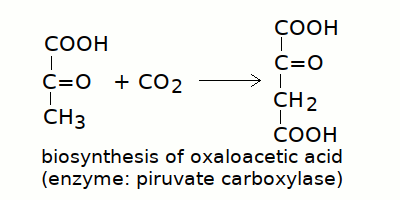
In the absence of glucose, piruvate is produced to a reduced extent, only from the metabolism of glycogenic aminoacids; as a consequence the mitochondrion is starved of oxaloacetic acid and the Krebs cycle slows down. Acetyl-CoA is thus produced in excess with respect to the ability of the Krebs cycle to oxidize it and accumulates in the mitochondrion. Acetyl-CoA polymerizes spontaneously to acetoacetyl-CoA and is finally released as acetoacetic acid. Two metabolytes of acetoacetic acid are also formed: acetone (by decarboxylation) and beta-hydroxy butanoic acid (by reduction). These three compounds, collectively called ketone bodies, accumulate in the blood and are ultimately excreted by the kidney (acetone, being volatile, is also excreted by the lung). 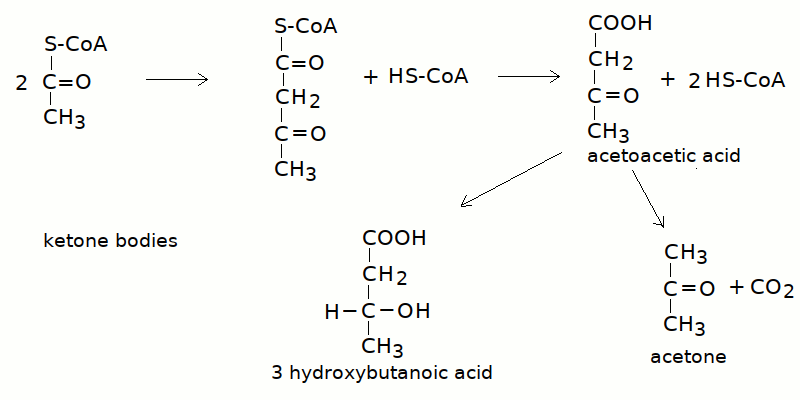
Since two ketone bodies are non volatile acids, they cause an important metabolic acidosis called diabetic ketoacidosis. Diabetic ketoacidosis is the most severe consequence of the disruption of cellular metabolism, and is the cause of death in untreated insulin dependent diabetes mellitus. By contrast, in type 2 diabetes, cellular usage of glucose is impaired but not abolished, and ketoacidosis, if present at all, is neither severe nor life treatening. Since glucose is lost in the urine and its metabolic role in the cell is played by aminoacids, protein waste occurs, with weight loss and negative nitrogen balance (more nitrogen is excreted than it is introduced with the diet). Hyperglycemia and increased glucose concentration in the extracellular fluids favour the proliferation of microorganisms and cause increased susceptibility to opportunistic infections (e.g. candidiasis). IDDM is associated to peripheral predominantly sensory neuropathy (causing pain), atherosclerosis of large arteries and microvascular damage. The pathogenetic mechanism of these complications has not been completely unravelled. These complications occur on a chronic basis in insulin-treated patients (untreated patients did not survive enough to provide casistics), and probably depend on multiple factors. Minor hyperglycemia or scattered periods of hyperglycemia are common in insulin-treated diabetic patients; this occurs because excess insulin may cause hypoglycemic coma and thus the patient and the physician prefer to use it careully. Over long times, frequently repeated minor episodes of hyperglycemia may damage the cell membranes, because of osmotic reasons and because of the reaction of glucose with membrane proteins. Ketone bodies may contribute to the damage, as well as the cellular metabolic inbalance. Vascular and neural damage may cause the diabetic ulcer of the extremities, which in turn is worsened by the increased susceptibility to infection. Another complication is damage of the retina which is to be ascribed to the combined effects of microvascular and neural damage. In untreated patients, accumulation of acetoacetic and beta-hydroxy butanoic acids cause a metabolic acidosis with reduced blood pH and compensatory hyperpnea (Kussmaul's breath) and hypocapnia (reduced CO2 and bicarbonate). Vomiting, and drowsiness are common symptoms of this condition; coma and death result if the blood pH drops to 7.1 or less. The cardinal symptoms of untreated IDDM, polyuria, polydipsia, acetone breath, and weight loss in an infant or adolescent should indicate dedicated laboratory tests. LATE ONSET DIABETES MELLITUS (NIDDM; Insulin Resistent Diabetes Mellitus) accounts for the great majority of the cases of DM, but is less severe than IDDM and has a better prognosis even if untreated or poorly treated. NIDDM (non Insulin Dependent DM) is a metabolic imbalance of the cells that causes a reduction of the insulin receptors on the cell surface. Hyperglycemia and glycosuria are present but the cells are not starved of glucose and ketoacidosis is uncommon. In extreme cases the blood glucose concentration may reach such high levels to cause hyperosmolarity and possibly coma and death. Since the hyperglycemia overstimulates insulin production the B cells of the Langerhans islets may undergo degeneration and the NIDDM may convert, with time, into IDDM albeit the mechanism of destruction of the insulin producing cells is not due to autoimmunity. NIDDM is associated to obesity and hypertension, and causes renal damage (because of glycosuria) and atherosclerosis of large arteries; microangiopathy is neither common nor prominent. Peripheral neuropathy, and diabetic ulcers of the extremity may appear in chronic poorly treated cases. The main differences between IDDM and NIDDM are summarized in the Table below.
DIABETES ASSOCIATED WITH OTHER ENDOCRINE DISEASES: other hormones participate to the control of glycemia and imbalances in their production (e.g. by hormone producing adenomas of the gland tissue) may cause hyperglycemia and glycosuria. Usually there are other symptoms associated with the specific hormone involved and no metabolic symptoms of diabetes (e.g. no ketoacidosis). The principal hormone that may cause a diabets-like syndrome is cortisol (and in general all glycocorticoids). The syndrome is suspected because of polyuria, and confirmed by the finding of hyperglycemia and glycosuria; the hormone involved must then be searched for in the blood (usually resorting to immunological methods). Morphological and functional study of the involved gland (e.g. NMR, PET and scintigraphy) is the next step. Galactosemia is a genetic disease inherited as an autosomal recessive trait. It is caused by absence of the enzyme galactosyl-1-phosphate uridyl transferase, whose gene is located on chromosome 9. The incidence is 1 newborn / 80,000 births and the diagnosis is usually established early, given the large apport of galactose from the milk lactose. The symptoms appear a few weeks after birth with vomiting, reduced weight gain and jaundice. Treatment is dietary, by substituting the mother's milk with an artificial one in which lactose has been replaced with glucose. In the absence of treatment reduced growth and impaired IQ are unavoidable and the life expectancy is reduced. Diagnosis is suspected upon finding a non glucose sugar in the urine, which can be demonstrated to be lactose or lactose-1-phosphate; genetic demonstration of the defect offer definitive confirmation. Other, less common, inherited causes of galactosemia and galactosuria are the inherited deficiencies of the enzymes galactokinase or galactose epimerase. The prognosis is less severe than in the case of defect of galactosyl-1-phosphate uridyl transferase. Fructosuria is caused by genetically determined lack of fructokinase; the incidence is 1/130,000 and the disease is benign. Hereditary fructose intolerance is due to the genetically determined defect of the enzyme phosphofructoaldolase, inherited as an autosomal recessive trait. In some countries or populations this defect may be relatively common (e.g. its frequency is 1/20,000 births in Switzerland). Fructose-1-phosphate accumulates in the body tissues and in the blood and appears in the urine. Severe symptoms follow the ingestion of fructose or suchrose (table sugar); renal damage may occur. Treatment require the elimination of fructose from the diet (not easy given that suchrose, the fructose containing disaccharide, is almost ubiquitary in fruits). Pentosuria is a harmless metabolic disorder due to lack of the enzyme L-xylulose dehydrogenase, and associated to excretion of L-xylulose in the urine. In some populations it is very common (e.g. 1/2,500 births among american Jews). The only risk for the patient is that his condition is misdiagnosed (and treated) for IDDM. Hypoglycemic conditions may cause fainting and coma. They may be due to several causes and, if severe, may cause death: thus they may present as medical emergencies. The mst common causes of hypoglycemias are as follows: Excess insulin administration in an IDDM patient, a quite common condition, may be life treatening and requires prompt diagnosis (caution! hypoglycemic coma needs to be distingushed from the two other possible types of coma in the diabetic patients: ketoacidosis and hyperosmotic). Insulin secreting adenoma of the Langerhans islets. Some hereditary diseases of carbohydrate metabolism: e.g. hereditary fructose intolerance, galactosemia, soem types of glycogenosis (see below). Deficiency of hormones causing increase of glycemia: glucagon, cortisol, growth hormone, hypothyroidism. Inherited defects of glucose metabolism are extremely rare (< 1:50,000). The single most important one is Pyruvate dehydrogenase complex deficiency (PDCD). Symptoms are poorly specific symptoms (lethargy, poor feeding, tachypnea, neurological disturbances), and are exacerbated by high carbohydrate intake. The disease is recessive and X-linked (only males are affected). Severity is variable; in severe cases prognosis is poor. Laboratory diagnosis relies on the demonstration of increased piruvate and lactate levels in the urine, blood and Cerebro Spinal Fluid (CSF). Alanine is also increased because of transamination of piruvate. Gene sequencing confirms the diagnosis. Glycogen storage disease (glycogenoses) are a group of (usually severe) genetic diseases of glycogen metabolism, causing accumulation of glycogen in several types of cells. A summary of the principal glycogeneoses is reported in the table below.
Home of this course |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||