PRIMA FACOLTA' DI MEDICINA E CHIRURGIA - CLM "B"
SAPIENZA UNIVERSITA' DI ROMA
A causa della sua limitata elettronegativita' il carbonio e' l'unico elemento della tavola periodica i cui atomi possono combinarsi tra loro senza limitazione di numero, generando molecole molto grandi e complesse. L'oggetto di studio della chimica organica sono queste molecole, i cui componenti principali, oltre al carbonio, sono l'idrogeno, l'azoto, l'ossigeno e talvolta anche lo zolfo ed il fosforo. Le molecole organiche sono di recente o remota origine biologica anche se possono essere prodotte artificialmente in laboratorio.
Gli idrocarburi sono i composti binari di carbonio e idrogeno e pertanto hanno la formula generale CnHm. Sono divisi nelle due grandi classi degli ALIFATICI e degli AROMATICI.
Gli idrocarburi ALIFATICI sono a loro volta divisi nelle seguenti classi:
ALCANI
ALCHENI
ALCHINI
POLIENI
CICLOALCANI
CICLOALCHENI
GLI ALCANI. Gli alcani presentano la formula generale CnH2n+2 con n variabile tra 1 e 70. Tutti gli atomi di carbonio hanno ibridazione sp3 e geometria tetraedrica e le molecole si presentano come lunghe catene carboniose, lineari o ramificate:
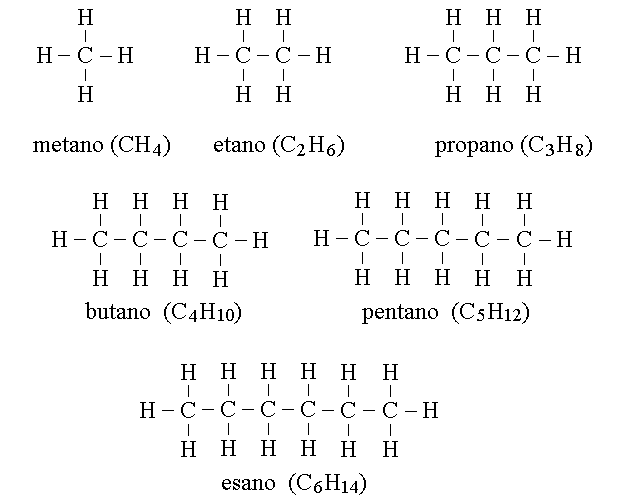
FORMULE RAZIONALI (O INTELLIGENTI). Nella chimica organica si fa spesso ricorso ad una notazione diversa dalla formula bruta (l'elenco degli atomi presenti nel composto) e dalla formula di struttura (la rappresentazione dettagliata di tutti gli atomi e tutti i legami), chiamata formula razionale. La formula razionale utilizza la notazione bruta per quei gruppi di atomi sulla cui disposizione non vi possono essere dubbi, e la notazione di struttura laddove sia essenziale. Ad esempio:
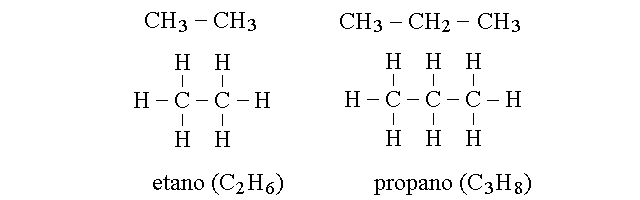
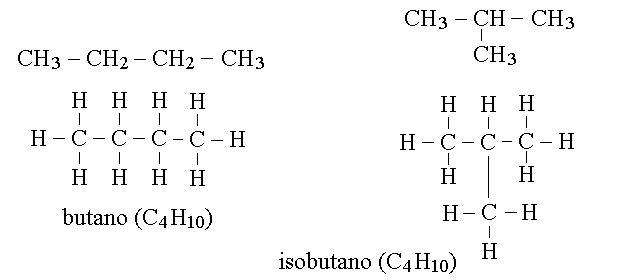
ISOMERIA NEGLI ALCANI. Fino al propano non abbiamo dubbi su come possano essere disposti gli atomi nella molecola; ma dell'idrocarburo successivo, il butano (C4H10), esistono due forme distinte con la stessa formula bruta; e di quello ancora successivo, il pentano (C5H12), ne esistono tre. Due o piu' molecole diverse che condividono la stessa formula bruta si definiscono isomeri. E' facile verificare che i due isomeri del butano (isomeri di costituzione)condividono la stessa formula bruta ma rappresentano due molecole diverse e non convertibili l'una nell'altra:
Gli alcani ammettono almeno altri due tipi di isomerie, chiamati isomeria di posizione e relativa alla posizione delle ramificazioni della catena principale di atomi di carbonio, e isomeria di conformazione, relativa alla rotazione degli atomi di carbonio rispetto al legame che li unisce. Entrambi saranno descritti meglio piu' avanti.
NUMERAZIONE DEGLI ATOMI DI CARBONIO. Gli atomi delle moelcole di carbonio sono numerati in modo tale che ciascuno sia identificato, secondo le regole seguenti (dettate dalla IUPAC, International Union for Pure and Applied Chemistry):
1) si individua la catena lineare ininterrotta (o ciclica) piu' lunga possibile, indipendentemente da come la molecola e' disegnata (infatti le curve che la catena carboniosa fa sul pezzo di carta non sono presenti nella realta', che prevede, per gli alcani, angoli di legame tutti uguali a 109,5 gradi).
2) si prova a numerare gli atomi di carbonio da ciascuna delle estremita' della catena cosi' individuata e si sceglie come inizio quella estremita' che assegna ai carboni legati ai sostituenti (atomi o gruppi diversi da H) il numero piu' basso.
3) si assegna alla molecola il nome dell'idrocarburo con lo stesso numero degli atomi di carbonio della catena trovata al punto 1, e si nominano i sostituenti di conseguenza.
Come esempio di questa procedura consideriamo la molecola riportata qui sotto, chiamata volgarmente isopentano. La catena carboniosa piu' lunga ha evidentemente 4 atomi di C (come il butano) e presenta un solo carbonio sostituito (che porta un gruppo CH3 - metile - al posto di un H). Comunque si provi a identificare e numerare la catena di atomi di C si trovera' che il numero piu' basso che si puo' assegnare al carbonio sostituito e' 2, come nella figura. Il nome IUPAC del composto e' quindi 2-metil butano.
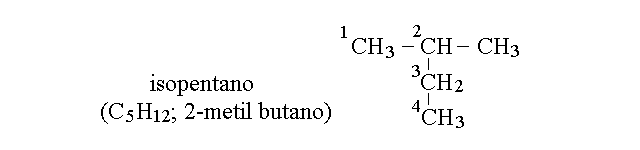
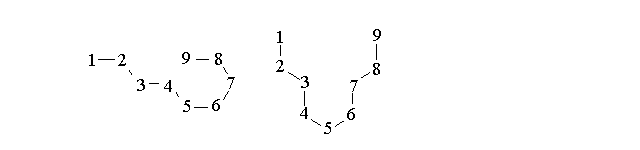
E' importante notare che a causa dell'ibridazione sp3, l'angolo di legame dei carboni negli idrocarburi e' sempre di 109,5 gradi e le eventuali "curve" della catena di atomi di carbonio disegnata sulla carta sono irrilevanti. Nella realta' ogni legame ruota su se stesso, mantenendo gli angoli di legame costanti, e le molecole si contorcono e si raddrizzano continuamente. Ad esempio l'alcano lineare a 9 atomi di carbonio (ennano) puo' assumere moltissime conformazioni spaziali due delle quali sono schematicamente rappresentate nella figura qui sotto. E' molto utile scaricare dalla rete un programma di visualizzazione molecolare e visualizzare le forme delle molecole (sebbene statiche) sul proprio computer: si utilizzino i links nella pagina della didattica.
CARBONIO PRIMARIO, SECONDARIO, TERZIARIO E QUATERNARIO. Un atomo di carbonio legato ad un solo altro atomo si definisce primario; ad esempio sono primari i due atomi di carbonio dell'etano (CH3-CH3) ed i carboni 1 e 3 del propano (CH3-CH2-CH3). Un atomo di carbonio legato ad altri due atomi di carbonio si definisce invece secondario; ne e' un esempio il C2 del propano. Quando un atomo di carbonio e' legato ad altri tre lo si definisce terziario, come nel caso del C2 del2-metil propano (isobutano), e quando e' legato ad altri quattro lo si definisce quaternario, come nel caso del C2 del 2,2 dimetil propano (neopentano).
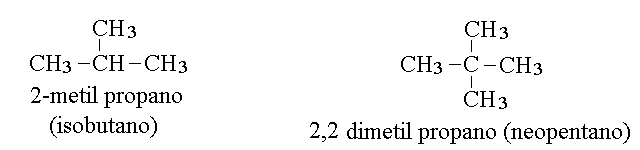
Il doppio legame carbonio-carbonio, caratteristico degli alcheni (vedi oltre) e' considerato come due legami e quindi i carboni dell'etene si considerano entrambi secondari; pero' in questo caso la classificazione non ha molta importanza perche' in genere nelle reazioni chimiche il doppio legame si rompe e quindi gli atomi di C degli alcheni possono cambiare categoria.
GLI ALCHENI. Gli alcheni sono simili agli alcani ma presentano un (solo) doppio legame nella catena di atomi di carbonio; i due atomi legati col doppio legame presentano l'ibridazione sp2 e la geometria trigonale planare con angoli di legame di 120 gradi; tutti gli altri presentano l'ibridazione sp3 e la geometria tetraedrica. La formula generale degli alcheni e' CnH2n. Il nome di ciascun alchene e' lo stesso dell'alcano con ugual numero di atomi di C, ma con la desinenza variata in -ene.
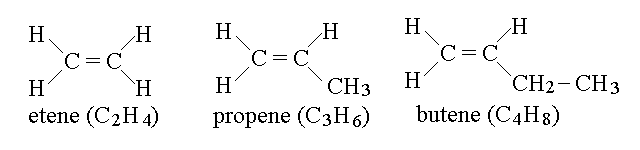
ISOMERIA NEGLI ALCHENI. Gli alcheni danno l'isomeria di costituzione, come gli alcani; si confrontino nella figura qui sotto l'1-butene e l'isobutene. Gli alcheni danno inoltre altri due tipi di isomeria: di posizione e geometrica. La prima e' relativa alla posizione del doppio legame ed e' presente in tutti gli alcheni cche abbiano almeno 4 atomi di carbonio: ci sono infatti almeno due possibili posizioni del doppio legame nel butene che configurano l'1-butene e il 2-butene. L'isomeria di posizione puo' interessare anche altre caratteristiche della molecola (ad es. per la posizione di una eventuale ramificazione, possibile anche negli alcani; si veda piu' avanti, classificazione delle isomerie). Quando il doppio legame assume una posizione interna nella molecola (cioe' nel nostro esempio nel caso del 2-butene), sono possibili due isomeri distinti a seconda che la catena carboniosa legata al secondo carbonio si trovi dalla stessa parte di quella legata al primo carbonio (forma cis) o dalla parte opposta (forma trans). L'isomeria geometrica e' possibile solo in presenza del doppio legame ed e' dovuta al fatto che il doppio legame impedisce la rotazione di un atomo di C rispetto all'altro: cioe' la conversione dell'isomero cis a quello trans richiede la rottura e la riformazione dell'orbitale di legame π.
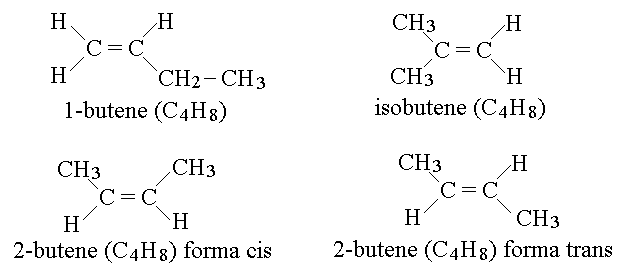
RICONOSCERE L'IBRIDAZIONE DEL CARBONIO (E DI QUALUNQUE ALTRO ATOMO). Per riconoscere l'ibridazione di ciascun atomo di carbonio, e di conseguenza la geometria e gli angoli di legame, in ogni molecola si segue questa regola: l'ibridazione e' sempre la piu' alta possibile, compatibilmente con gli orbitali di legame π. Infatti l'orbitale di legame di tipo π puo' essere formato (nel carbonio) soltanto a partire da orbitali atomici di tipo p non ibridizzati. Si deve ricordare che ogni doppio legame e' formato da un orbitale di tipo σ e uno di tipo π ogni triplo legame da un orbitale di tipo σ e due di tipo π. Pertanto: un carbonio che non forma doppi legami (come nel metano o nell'etano) ha ibridazione sp3; un carbonio che forma un solo doppio legame (come nell'etene) ha ibridazione sp2 e mantiene un orbitale atomico p non ibridato necessario per formare l'unico orbitale di legame π a cui partecipa (gli altri sono orbitali di legame di tipo σ); un carbonio che forma due doppi legami (come nell'anidride carbonica, O=C=O) o un triplo legame (come nell'etino o nell'acido cianidrico) ha ibridazione sp.
GLI ALCHINI. Gli alchini sono simili agli alcani ma presentano un (solo) triplo legame nella molecola. I due atomi legati col triplo legame presentano l'ibridazione sp e la geometria lineare con angolo di legame di 180 gradi; tutti gli altri presentano l'ibridazione sp3 e la geometria tetraedrica. La formula generale degli alchini e' CnH2n-2. Il nome di ciascun alciene e' lo stesso dell'alcano con ugual numero di atomi di C, ma con la desinenza variata in -ino. Dal butino in poi, gli alchini presentano l'isomeria di posizione come gli alcheni.
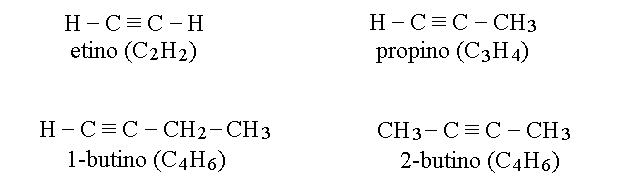
I POLIENI. I composti che possiedono due o piu' doppi legami si chiamano dieni, trieni ed infine polieni.
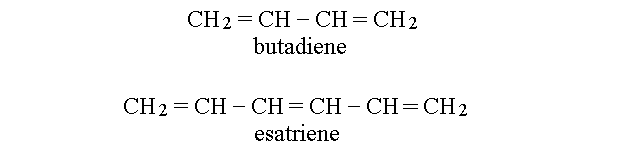
Quando in un poliene si alternano legami semplici e doppi, tutti i carboni presentano l'ibridazione sp2 ed e' possibile la delocalizzazione degli orbitali di legame di tipo π. Una struttura cosi' costituita si definisce "a doppi legami coniugati" ed e' presente in molte molecole di importanza biologica.
I CICLOALCANI. Esistono molecole di idrocarburi saturi, con carboni ibridati sp3 che si chiudono su se stessi a formare anelli; questi composti sono chiamati cicloalcani. La formula generale dei cicloalcani e' CnH2n e pertanto quetsi composti sono isomeri di funzione degli alcheni che presentano lo stesso numero di atomi di carbonio. Ad esempio il ciclobutano, la cui formula di struttura e' riportata nella figura qui sotto, presenta la formula bruta C4H8 ed e' pertanto un isomero del butene.
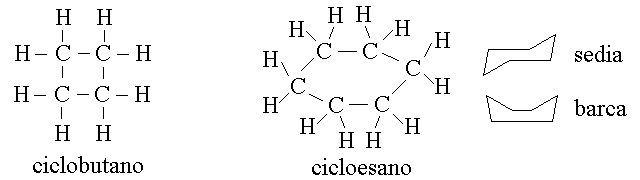
L'ibridazione sp3 caratteristica dei cicloalcani e di alcuni carboni della molecola dei cicloalcheni non e' compatibile con una geometria planare; pertanto i ciloalcani (e i cicloalcheni) con piu' di tre atomi di carbonio hanno forme caratteristiche; ad esempio il cicloesano puo' assumere due configurazioni distinte dette "a sedia" e "a barca". Queste configurazioni non sono planari (gli atomi di C si trovano su due piani distinti) ma non sono neppure distorte, perche' consentono angoli di legame di 109,5 gradi, come richiesto dall'ibridazione sp3.
I CICLOALCHENI E I POLIENI CICLICI. Come gli alcani anche gli alcheni e i polieni possono formare molecole cicliche. Ad esempio:
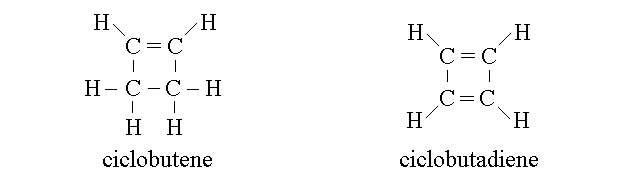
I cicloalcheni e i polieni ciclici sono sempre molecole non planari e dalle geometrie distorte (cioe' gli angoli di legame effettivi sono diversi da quelli ottimali per l'ibridazione effettiva, sp2 o sp3, degli atomi di carbonio coinvolti). Ad esempio il ciclobutadiene ha una configurazioen "a sella" con gli atomi di carbonio disposti su due piani. C'e' una sola geometria non distorta possibile per i polieni ciclici, ed e' quella di un anello planare a sei atomi di carbonio con tre doppi legami coniugati; quando questa geometria si realizza pero' il composto considerato esce dalla categoria dei polieni ciclici per entrare in quella dei composti aromatici.
IDROCARBURI AROMATICI.
Gli idrocarburi aromatici assomigliano a polieni ciclici a sei atomi di carbonio con doppi legami coniugati (cioe' alternati a legami semplici); eccezionalmente ve ne sono a cinque o a sette. Tutti gli atomi di carbonio presentano ibridazione sp2 e geometria trigonale planare, con angoli di legame di 120 gradi, perfettamente compatibile con la forma geometrica dell'esagono regolare (cioe' la struttura molecolare e' non-distorta perche' gli angoli interni del poligono e quelli ottimali per l'ibridazione degli orbitali coincidono). La forma di esagono regolare, perfettamente planare, consente l'esistenza di ibridi di risonanza (strutture con gli atomi nelle stesse posizioni ma una diversa disposizione degli orbitali π) e la delocalizzazionedegli orbitali di legame π questo a sua volta conferisce grande stabilita' (cioe' scarsa reattivita') alla molecola. La delocalizzazione degli orbitali di legame π e' spesso rappresentata con un circoletto disegnato all'interno dell'anello a sei atomi.
L'idrocarburo aromatico piu' semplice e' il benzene, con un solo anello a sei atomi (formula bruta C6), ma ve ne sono a piu' anelli condensati: il naftalene, che ha due anelli (C10H8), gli isomeri antracene e fenantrene, a tre anelli (C14H10), etc. I composti aromatici rispettano la regola di Huckel, secondo la quale gli elettroni presenti negli orbitali di legame π delocalizzati sono 4n+2 dove n rappresenta il numero di anelli condensati (1 per il benzene, 2 per il naftalene, etc.).
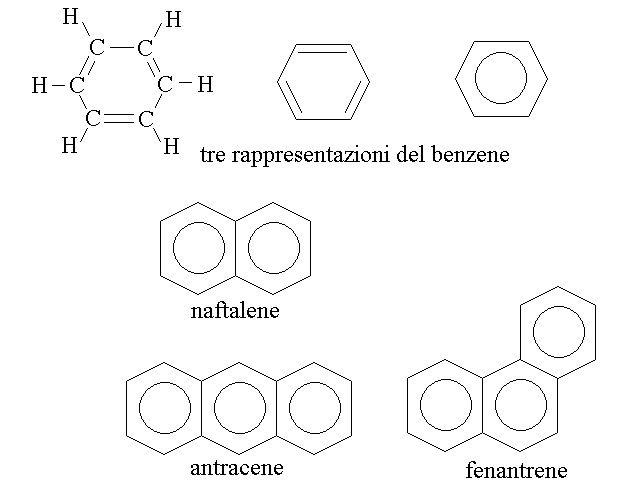
Gli idrocarburi aromatici possono presentare brevi catene alifatiche ai vertici dell'anello aromatico; ad esempio:
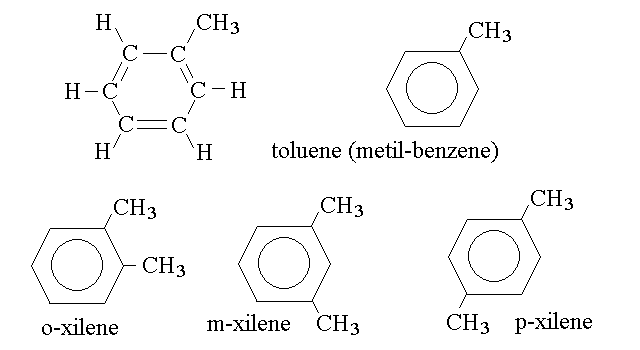
Si noti la nomenclatura delle posizioni relative dei due sostituenti sull'anello aromatico dello xilene: le posizioni contigue corrispondono alla dizione orto (orto- o o-xilene, 1,2-dimetil benzene); separate da un solo atomo di carbonio alla dizione meta (metaa- o m-xilene, 1,3-dimetil benzene); separate da due atomi di carbonio alla dizione para (para- o p-xilene, 1,4-dimetil benzene). Per ovvi motivi di simmetria le posizioni 1,5 e 1,6 non esistono (sono rispettivamente le 1,3 e 1,2 numerate male).
RESIDUI. Si definisce residuo o radicale un composto immaginario corrispondente ad un idrocarburo al quale sia stato sottratto un atomo di idrogeno. Questa operazione lascia sul carbonio un elettrone spaiato, estremamente reattivo. Il residuo non esiste in natura da solo, se non come intermedio instabile delle reazioni che avvengono con meccanismo radicalico; esiste invece in forma combinata con altri atomi o gruppi, in modo che la molecola completa, formata da residuo e gruppo, ha tutti elettroni appaiati. Il residuo e' una astrazione utile per descrivere i composti piu' complessi come se fossero formati da residui e gruppi combinati tra loro. Il nome del residuo e' lo stesso dell'idrocarburo da cui deriva ma con la desinenza cambiata in -ile; se lo si deve rappresentare da solo lo si indica con un puntino che rappresenta l'elettrone spaiato.
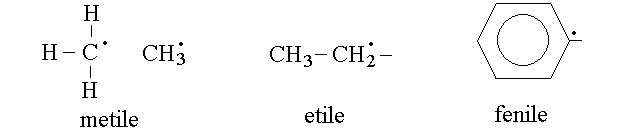
REAZIONI DEGLI ALCANI. La reazione caratteristica degli alcani e' la sostituzione di un atomo di idrogeno con un atomo diverso; ad esempio:
Il prodotto di reazione, ovviamente, non e' piu' un alcano (nell'esempio qui sopra, l'etano viene trasformato in cloroetano).
REAZIONI DEGLI ALCHENI E DEGLI ALCHINI. Negli alcheni e negli alchini, l'orbitale di legame di tipo π costituisce un punto debole della molecola ed e' facilmente attaccato dalle reazioni di addizione. Ad esempio:
L'addizione di idrogeno si chiama saturazione. Questa reazione e' possibile per gli alcheni e gli alchini, che di conseguenza si chiamano idrocarburi insaturi, ma non per gli alcani (idrocarburi saturi). Il prodotto della reazione di saturazione di un alchene e' un alcano; quello della reazione di un alchino e' un alchene (e, in eccesso di reagente, eventualmente un alcano). Indicando con X un riducente capace di donare atomi di idrogeno, la reazione di saturazione ha lo schema:
REAZIONI DEGLI AROMATICI. La delocalizzazione degli elettroni negli orbitali di legame π conferisce una grande stabilita' agli idrocarburi aromatici, grazie ad un forte contributo entropico. La eventuale reazione di addizione comporterebbe l'interruzione della delocalizzazione e quindi ha un contributo entropico negativo (sfavorevole). Di conseguenza gli aromatici danno piu' facilmente le reazioni di sostituzione (come gli alcani), che non interrompono l'anello aromatico, che quelle di addizione (come gli alcheni). Questa reattivita' e' assai caratteristica degli aromatici, che sono idrocarburi insaturi ma si comportano come se fossero saturi.
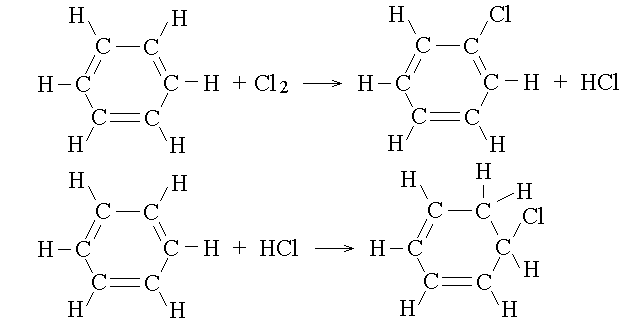
E' evidente dalla figura che la reazione di sostituzione (in alto) non altera l'ibridazione degli atomi di carbonio del benzene e la distribuzione dei doppi legami: il carattere aromatico dell'anello a sei atomi viene quindi conservato e gli orbitali π rimangono delocalizzati. Per contro in una eventuale reazione di addizione (in basso) due atomi di carbonio passerebbero dall'ibridazione sp2 alla sp3, ed un doppio legame sarebbe perduto. Questo avrebbe come conseguenza l'impossibilita' di delocalizzare i due orbitali π rimasti (infatti e' impossibile scrivere le formule degli ibridi di risonanza). Ad esempio si puo' osservare che il calore di idrogenazione del cicloesene e' di circa -29 kcal/mole e quello del cicloesadiene e' circa il doppio (-57 kcal/mole); per contro il calore di idrogenazione del benzene e' di circa -50 kcal/mole. Questo implica che i valori medi del calore di idrogenazione di ciascun doppio legame risultano -28,5 kcal/mole nei primi due idrocarburi e -16,7 nel benzene; ovvero che il benzene risulta piu' stabile di quanto si potrebbe calcolare dalla somma dei calori di idrogenzaione dei suoi doppi legami per circa 36 kcal/mole (28,5x3-50=35,5). Il guadagno di stabilita' conferito dalla natura aromatica non e' identico in tutti i composti aromatici.
I composti ternari del carbonio sono molto numerosi; per descriverli e classificarli si ricorre al seguente artificio: si immagina che questi composti siano costruiti unendo il residuo di un idrocarburo ad un gruppo funzionale (o funzione chimica) che puo' contenere o non contenere altri atomi di carbonio. Sia il residuo che il gruppo funzionale sono radicali, cioe' composti immaginari che presentano un elettrone spaiato, necessario per legarsi tra loro, e in genere non esistono come tali in natura se non nella forma di intermedi instabili di reazioni chimiche. Poiche' i residui degli idrocarburi sono stati descritti sopra, descriveremo in questa sezione i soli gruppi funzionali principali.
GRUPPI FUNZIONALI DELL'OSSIGENO.
I gruppi funzionali dell'ossigeno sono numerosi ed importanti.
GLI ALCOLI E I FENOLI. Il piu' semplice gruppo funzionale contenente ossigeno e' il gruppo ossidrile, -OH, presente negli alcoli e nei fenoli. Gli alcoli sono i derivati ossidrilici dei carboni alifatici, i fenoli i derivati ossidrilici dei carboni appartenenti ad anelli aromatici. Secondo la IUPAC, il nome di un alcol e' quello dell'idrocarburo corrispondente al quale viene aggiunta la desinenza -olo. Ad esempio l'alcol dell'etano ha formula CH3-CH2OH e nome etanolo; quello del propano ha formula CH3-CH2-CH2OH e nome propanolo; etc.
Un alcol e' definito primario se il gruppo funzionale -OH e' legato ad un carbonio primario (come in entrambi gli esempi riportati sopra). In un alcol secondario il gruppo ossidrile e' legato ad un carbonio secondario ed in un alcol terziario ad un carbonio terziario. Ovviamente non possono esistere alcoli quaternari.
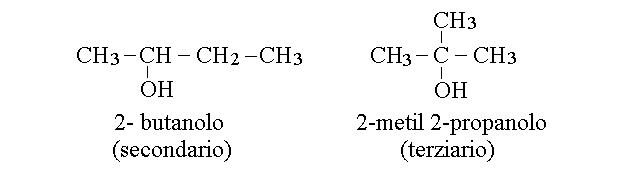
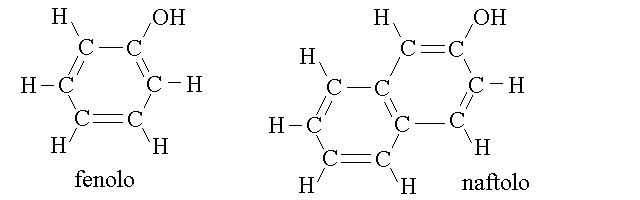
Fenolo e' sia un nome proprio che un nome comune. Come nome proprio indica l'alcol del benzene (considerato un alcol terziario); come nome comune indica invece qualunque aromatico nel quale un idrogeno legato ad un carbonio aromatico e' sostituito con un gruppo ossidrile. Pertanto e' corretto dire che il fenolo ed il naftolo sono due esempi di fenoli:
Gli alcoli alifatici non si comportano ne' da acidi ne' da basi; questo equivale a dire che la molecola non dissocia ne' H+ ne' OH-, o, se si preferisce, che i legami C-O e O-H negli alcoli sono solo debolmente polarizzati (infatti in acqua si rompono soltanto i legami ionici o fortemente polari). Si confrontino le seguenti molecole, nelle quali il legame piu' polare, che ha tendenza a rompersi quando il composto e' disciolto in acqua, e' indicato con una freccia:
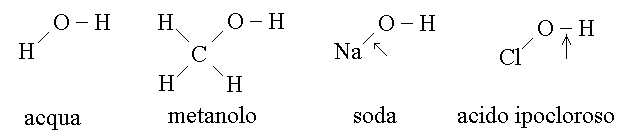
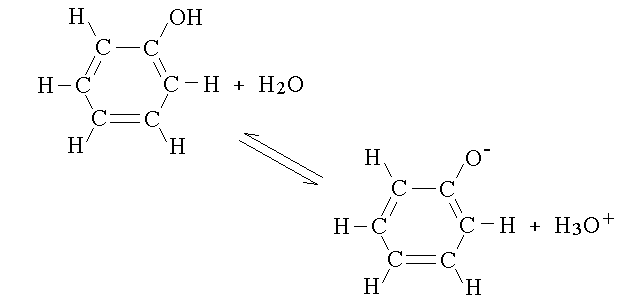
Per contro, a causa dell'elettronegativita' dell'anello aromatico, che ha una forte capacita' di attrarre gli elettroni nel sistema degli orbitali π delocalizzati, il legame tra O e H nel fenolo e' alquanto polarizzato e il composto si comporta come un acido debole con pKa = 9:
Gli alcoli possono essere considerati prodotti di ossidazione degli idrocarburi corrispondenti, immaginando o realizzando la reazione con un ossidante capace di cedere ossigeno quale l'acqua ossigenata:
In genere non e' facile arrestare la reazione a questo punto e convertire quantitativamente un idrocarburo in un alcol: in presenza di un eccesso di ossidante la reazione procede oltre producendo una miscela di composti con diversi gardi di ossidazione (ad es. aldeidi ed acidi carbossilici, vedi oltre).
Per dimostrare che la reazione che converte un idrocarburo in un alcol e' una ossidazione, calcoliamo il numero di ossidazione dei carboni dell'etano e dell'etanolo. Nell'etano i due carboni hanno ovviamente lo stesso numero di ossidazione e questo risulta essere -3; infatti ogni carbonio riceve un elettrone da ciascuno dei tre idrogeni ai quali e' legato (che hanno n.o. = +1), mentre il legame carbonio-carbonio e' omopolare e non da luogo a scambio di elettroni. Nell'etanolo il primo atomo di carbonio riceve due elettroni da ciascuno degli atomi di idrogeno a cui e' legato e ne cede uno all'ossigeno (che ha n.o. = -2 e prende il secondo elettrone dall'idrogeno cui e' legato); il numero di ossidazione del C1 dell'etanolo risulta pertanto -2+1=-1. Il secondo atomo di carbonio dell'etanolo compare in un gruppo CH3 ed il suo numero di ossidazione e' lo steso gia' calcolato per i carboni dell'etano. Nella conversione dell'etano ad etanolo, pertanto, il C1 passa da un numero di ossidazione piu' basso (-3) ad uno piu' alto (-1) come e' caratteristico delle ossidazioni. La specie che si riduce nella reazione considerata e' invece l'ossigeno che passa da n.o.=-1 (nell'acqua ossigenata) a n.o.=-2 (nell'etanolo e nell'acqua).
LE ALDEIDI E I CHETONI. Le aldeidi ed i chetoni presentano il gruppo funzionale carbonile e possono essere ottenuti mediante ossidazione dei corrispondenti alcoli primari (le aldeidi) o secondari (i chetoni). Coerentemente, la riduzione dell'aldeide restituisce l'alcol primario corrispondente, quella del chetone l'alcol secondario corrispondente. Gli alcoli terziari non danno reazioni di ossidazione. La formula del gruppo carbonilico e':
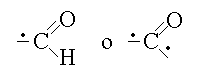
Il nome IUPAC dell'aldeide si ottiene aggiungendo la desinenza -ale al nome dell'idrocarburo con lo stesso numero di atomi di carbonio; quello del chetone aggiungendo la desinenza -one. Il numero di ossidazione del carbonio del gruppo aldeidico e' +1; quello del carbonio del gruppo chetonico +2.
La reazione di ossidazione necessaria per convertire un alcol primario in una aldeide richiede di sottrarre due atomi di idrogeno (o meglio due ioni idrogeno e due elettroni) al carbonio, ed il nome di aldeide storicamente deriva dalla contrazione di "alcol de-idrogenato". Conseguentemente l'ossidante adatto deve potersi combinare con atomi di idrogeno ed elettroni:
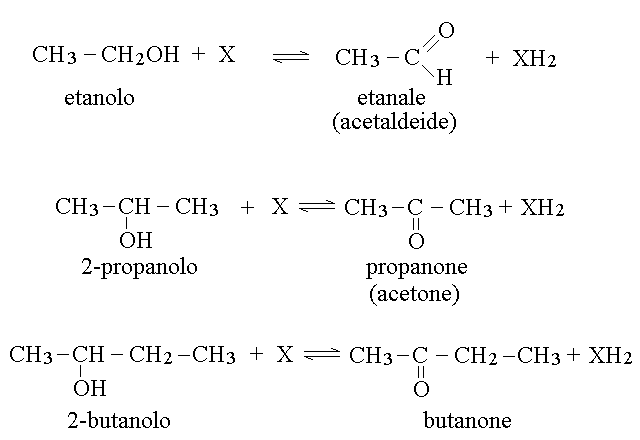
Il gruppo carbonilico presenta un atomo di carbonio con ibridazione sp2 e geometria trigonale planare; pertanto nell'ossidazione da alcol ad aldeide o da alcol a chetone si ha anche un cambiamento dell'ibridaizone del carbonio da sp3 a sp2.
CHINONI E IDROCHINONI. Poiche' l'alcol terziario non da' ossidazione, non la danno neppure i fenoli, con una eccezione: quella di una classe di composti chiamati idrochinoni (nella forma ridotta, alcolica) e chinoni (nella forma ossidata, chetonica). I chinoni sono trasportatori di elettroni, molto importanti in biologia:
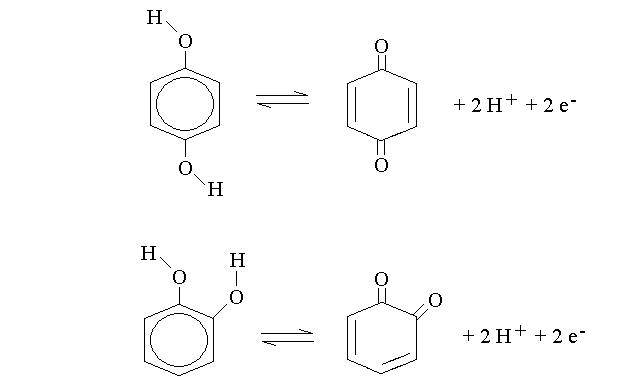
Un vero idrochinone deve contenere almeno due gruppi idrossilici legati ad un idrocarburo aromatico e questi devono trovarsi in posizioni tali da consentire che gli atomi di carbonio ai quali sono legati mantengano l'ibridazione sp2 sia nella forma ridotta (idrochinone) che in quella ossidata (chinone). Ad esempio vi sono due possibili idrochinoni del benzene (riportati nella figura qui sopra) con sostituzioni in posizioni orto e para, mentre non e' un vero idrochinone l'1,3 di-idrossi benzene (cioe' il composto aromatico che presenta sostituzioni in posizioni meta), perche' non e' possibile scrivere una formula ossidata che mantenga per tutti gli atomi di carbonio di questo composto l'ibridazione sp2 (provare per credere!). Nell'idrochinone il sistema di orbitali π delocalizzati copre l'anello a sei atomi ed esclude gli atomi di ossigeno; nel chinone, invece, la delocalizzazione si estende anche agli orbitali π del doppio legame C=O.
GLI ACIDI CARBOSSILICI. Il gruppo funzionale carbossile caratterizza gli acidi carbossilici. Le formule del gruppo carbossilico e di alcuni acidi carbossilici sono riportate qui sotto.
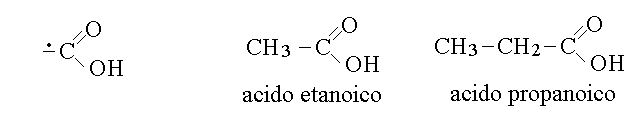
Il nome degli acidi carbossilici, secondo la IUPAC si corstuisce aggiungendo la desinenza -oico al nome dell'idrocarburo con lo stesso numero di atomi di carbonio. Per ragioni storiche pero' molti acidi carbossilici hanno nomi convenzionali, non IUPAC, e questi sono spesso piu' usati (ad es. l'acido metanoico si chiama formico e l'etanoico si chiama acetico).
L'acido carbossilico e' il prodotto dell'ossidazione dell'aldeide corrispondente:
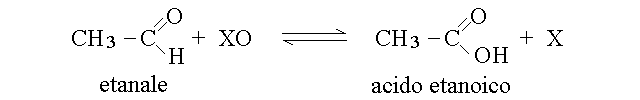
(dove XO rappresenta un generico ossidante e X la sua forma ridotta). I chetoni, naturalmente, non possono ossdiarsi in questo modo. Il numero di ossidazione del carbonio nel gruppo carbossilico e' +3.
Gli acidi carbossilici sono acidi deboli con pKa compreso tra 3 e 6; ad es.:
CH3-COOH + H2O <==> CH3-COO- + H3O+
GRUPPI FUNZIONALI DELL'AZOTO.
I gruppi funzionali dell'azoto sono due: il gruppo aminico o amminico (che assomiglia all'ossidrile degli alcoli) e quello amidico o ammidico (che assomiglia all'OH degli acidi carbossilici)
LE AMINE O AMMINE. Il gruppo aminico primario ha la formula -NH2. Esistono inoltre i gruppi aminici secondario (-NH-), terziario (-N<), e quaternario. L'amina secondaria e' talvolta chiamata imina. Ad esempio:
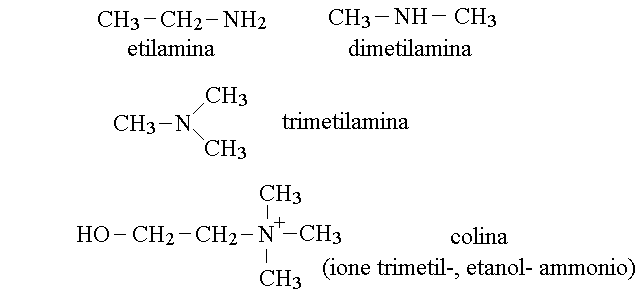
Le ammine quaternarie sono obbliagtoriamente ioni positivi: infatti il gruppo aminico quaternario e' uno ione ammonio legato a quattro residui organici, nel quale e' presente un atomo di azoto che ha perduto un elettrone ed ha quindi acquisito la configurazione elettronica dell'elemento che lo precede nella tavola periodica (il carbonio). Quando una amina quaternaria viene isolata, la si ottiene sempre in presenza di uno ione negativo, come sale (ad es. il cloruro di colina) o come idrossido. Uno ione ammonio quaternario molto importante per la fisiologia e' la colina (riportata in figura).
Le amine sono derivati dell'ammoniaca e l'azoto presenta ibridazione sp3 e geometria tetraedrica, come nell'ammoniaca; inoltre tutte le amine sono basi deboli con pKb compreso all'incirca tra 4 e 6. Per contro lo ione ammonio quaternario non e' ne' una base debole, ne' l'acido coniugato di una base debole e si comporta in soluzione come uno ione spettatore.
E' importante notare che mentre un alcol e' primario, secondario o terziario in relazione all'atomo di carbonio al quale e' legato l'ossigeno, una amina e' primaria, secondaria o terziaria in funzione dei legami dell'azoto. Di conseguenza una amina secondaria come la dimetilamina non assomiglia ad un alcol secondario come il 2-propanolo (guardare le formule!) ma semmai all'etere dimetilico (vedi oltre).
Esiste un tipo particolare di amina secondaria, anch'essa chiamata imina che scambia due legami con lo stesso atomo di carbonio (forma cioe' un doppio legame e assomiglia ad una aldeide o chetone); ad esempio la metil-imina (CH2=NH). Le imine sono in genere composti instabili in acqua e possono essere preparate in solventi organici. Tra le varie reazioni che danno con l'acqua, e che le trasformano, la principale e' l'ossidazione ad aldeide o chetone:
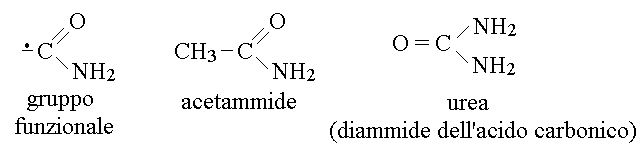
LE AMIDI O AMMIDI. Il gruppo funzionale amidico ha la formula -CONH2, come nelle formule seguenti:
La struttura del gruppo amidico consente la delocalizzazione dell'orbitale di legame π sui tre atomi C, O ed N e questo impone la geometria sp2 per l'atomo di azoto ed il parziale impegno del doppietto spaiato dell'azoto nel sistema delocalizzato O-C-N. Come conseguenza le amidi non hanno carattere basico. Per raffigurarsi questa struttura si possono scrivere, oltre alla forma delocalizzata (la piu' realistica), anche i suoi ibridi di risonanza:
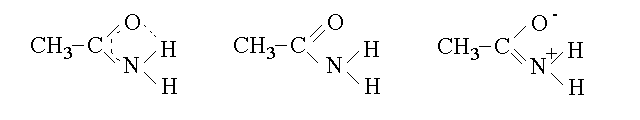
L'esame degli ibridi di risonanza del gruppo amidico ci chiarisce un interessante aspetto del mancato comportamento basico di questi composti: infatti uno dei due ibridi di risonanza si presenta come una imina protonata e ci consente di dire che l'azoto soddisfa la sua basicita' grazie al semplice riarrangiamento degli elettroni, senza bisogno di un donatore esterno di ioni idrogeno (acido); ovvero l'amide si "dona" il suo protone da se' stessa.
Il comportamento neutro, anziche' basico, delle amidi ha rilievo in fisiologia. Infatti gli organismi animali assumono azoto nella dieta e questo elemento non puo' essere ossidato (come accade per il carbonio che viene trasformato in CO2), ma deve essere eliminato per via urinaria nella forma dell'ammoniaca o di un derivato aminico o amidico. I pesci eliminano l'azoto come ammoniaca e sono definiti animali ammoniotelici; poiche' l'ammoniaca e' basica, la sua eliminazione richiede di diluirla nell'urina, un problema minore per animali che vivono in acqua. I mammiferi e molti vertebreti terrestri sono invece ureotelici cioe' convertono l'azoto in urea ed eliminano questa nelle urine. L'urea puo' essere eliminata a concentrazioni relativamente elevate perche' non e' basica e quindi non danneggia il sistema urinario. Il sangue dell'uomo contiene urea, prodotta al solo scopo di eliminare l'azoto, e la sua concentrazione viene determinata degradando la molecola e misurando l'azoto liberato; il valore medio dell'azotemia nella popolazione sana risulta pari a circa 30-40 mg/100 ml (corrispondenti a circa 80-90 mg di urea/100 ml).
GRUPPI FUNZIONALI DELLO ZOLFO.
In pratica, l'unico gruppo funzionale importante dello zolfo e' il tioalcol o mercaptano, che somiglia all'alcol, ed ha la formula -SH. Si deve ricordare che lo zolfo e' l'omologo superiore dell'ossigeno, cioe' l'elemento dello stesso gruppo (il sesto) ma appartenente al periodo successivo (il terzo invece del secondo; controllare sulla tavola periodica) e pertanto ha la stessa configurazione elettronica esterna e reattivita' simile. Un esempio di tioalcol e' l'etantiolo (o mercaptoetanolo), che ha la formula CH3-CH2-SH.
A differenza degli alcoli, i tioalcoli sono acidi deboli con pKa di circa 9:
GRUPPI FUNZIONALI DERIVANTI DALLA REAZIONI DI ALTRI GRUPPI FUNZIONALI.
I gruppi funzionali studiati fin qui esauriscono piu' o meno tutti i composti piu' frequenti nella chimica biologica, ma non considerano quei possibili ulteriori gruppi funzionali piu' complessi che possono originarsi da reazioni di quelli semplici tra loro. I gruppi piu' reattivi sono quelli che contengono l'ossigeno ed e' da questi che conviene cominciare.
IL GRUPPO ETERE. L'etere e' il prodotto della combinazione di due alcoli, con eliminazione di una molecola d'acqua:
Ci sono alcune osservazioni da fare su questa reazione:
1) la reazione non e' una ossido-riduzione; infatti il numero di ossidazione del carbonio del metanolo (l'esempio riportato qui sopra) e' -2 e quello di ciascuno dei due carboni del dimetiletere e' ancora -2; l'idrogeno e' sempre +1 e l'ossigeno sempre -2.
2) la reazione passa attraverso la sottrazione dell'idrogeno di un gruppo -OH da parte dell'ossigeno dell'altro gruppo OH.
3) in assenza di acqua e' favorita la formazione dell'etere; invece in presenza di acqua e' favorita la reazione inversa di dissociazione dell'etere in due alcoli.
ACETALI E CHETALI. Il semiacetale e l'acetale derivano dalla reazione di uno o due alcoli con una aldeide; il semichetale e il chetale derivano dalla reazione di uno o due alcoli con un chetone. Ad esempio:
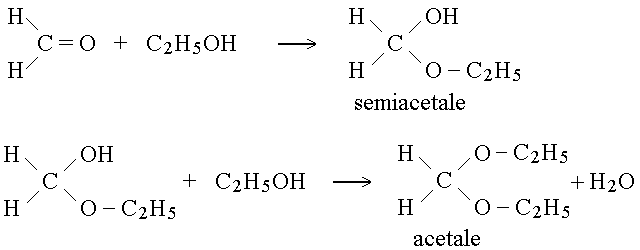
Anche su questa reazione ci sono alcune osservazioni da fare:
1) neppure questa reazione e' una ossido-riduzione
2) il carbonio carbonilico dell'aldeide o del chetone presenta l'ibridazione sp2 e la geometria trigonale planare mentre quello del semiacetale, acetale, semichetale o chetale presenta l'ibridazione sp3 e la geometria tetraedrica. Si osserva quindi nella reazione un cambio della geometria del carbonio.
3) in conseguenza del precedente punto (2) un carbonio che non presentava forme isomeriche (dell'aldeide o del chetone)viene trasformato in un carbonio che potrebbe presentare il fenomeno dell'isomeria ottica (vedi oltre).
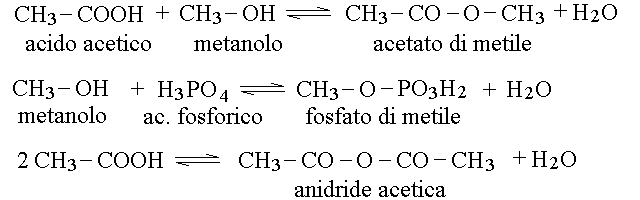
ESTERI E ANIDRIDI. L'estere e' il prodotto della reazione di un alcol con una acido, con eliminazione di una molecola d'acqua. Se l'acido e' un acido carbossilico, l'estere ottenuto e' a sua volta un estere carbossilico; ma questa non e' una condizione necessaria e qualunque ossiacido puo reagire con un alcol per produrre un estere (ad esempio hanno grande importanza in biologia gli esteri organici dell'acido fosforico). L'anidride e' un acido deidratato, cioe' un acido a cui e' stata sottratta una molecola d'acqua. A second adella formula dell'acido, la deidratazione puo' richiedere una sola molecola di acido (come accade ad esempio nella coppia acido carbonico-anidride carbonica)accompagnarsi alla combinazione di due molecole di acido tra loro (come accade nella coppia acido acetico-anidride acetica). Ne' la formazione dell'estere ne' quella dell'anidride comportano ossido-riduzione, come si puo' facilmente verificare calcolando il numero di ossidazione degli atomi di carbonio.
REAZIONI DEI TIOLI. Come gli alcoli corrispondenti, anche i tioli possono dare tioeteri e tioesteri; inoltre il gruppo tiolico puo' dare una reazione di ossido-riduzione che l'alcol non puo' dare, il cui prodotto e' il disolfuro. Esempi di formule razionali per questi composti sono i seguenti:
CH3-CH2-S-CH2-CH3 (dietiltioetere)
CH3-CO-S-CH2-CH3 (tioacetato di etile, un tioestere)
CH3-CH2-S-S-CH2-CH3 (disolfuro dell'etantiolo)
BASI DI SCHIFF. La base di Schiff e' una amina terziaria piuttosto peculiare, che si ottiene dalla reazione (reversibile) di una amina primaria con una aldeide; per riduzione puo' essere trasformata (irreversibilmente) in una amina secondaria.
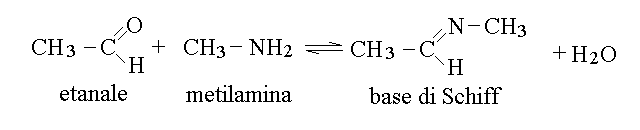
COMPOSTI ETEROCICLICI. Gli eterociclici sono composti ciclici, aromatici o alifatici, nel cui anello sono presenti atomi diversi dal carbonio. Tra gli etrociclici alifatici importanti nominiamo il tetraidrofurano, il tetraidropirano, il pirano e la piperidina:
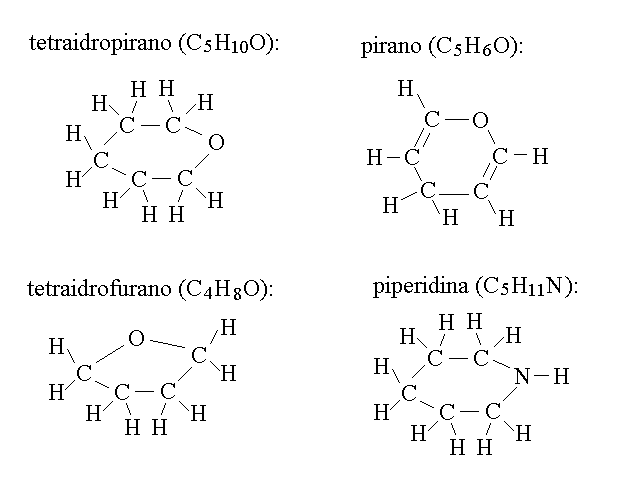
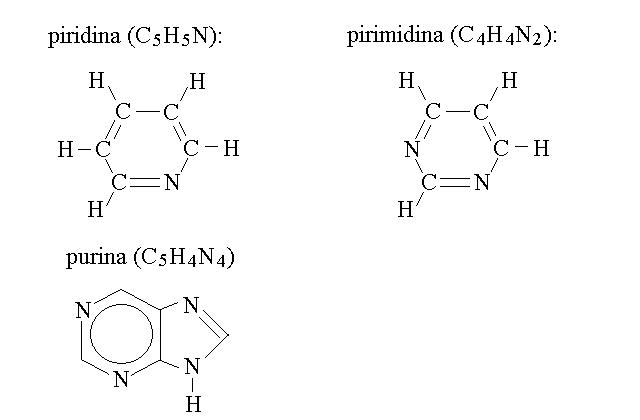
Tra gli eterociclici a carattere aromatico ricordiamo la piridina, la pirimidina e la purina:
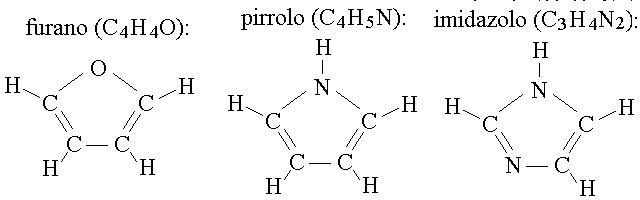
Esistono infine alcuni composti eterociclici con carattere aromatico, nei quali l'anello e' pentatomico. In questi casi la regola di Huckel e' rispettata perche' uno dei 5 atomi (ad es. l'azoto) fornisce una coppia di elettroni che si sommano ai 4 elettroni forniti dai 4 atomi di carbonio. I piu' importanti dal punto di vista della biochimica sono il furano, il pirrolo e l'imidazolo:
Non e' necessario conoscere i meccanismi di reazione di tutte le reazioni dei gruppi funzionali studiati; e' pero' importante avere almeno un'idea dei piu' comuni meccanismi delle reazioni della chimica organica.
Il principale meccanismo delle reazioni di sostituzione e' radicalico: passa cioe' attraverso la formazione di radicali liberi (atomi o composti che presentano un elettrone spaiato, molto reattivi). I meccanismi radicalici hanno elevate energie di attivazione iniziali (cioe' e' difficile iniziare la reazione e produrre il primo radicale; questo richiede spesso irraggiamento X o UV); pero' una volta che questo si e' formato la reazione si automantiene perche' altri radicali vengono formati dalla reazione del primo. Un esempio di sostituzione radicalica e' dato dall'alogenazione degli alcani, come nello schema seguente, nel quale le freccioline indicano la separazione degli elettroni di legame:
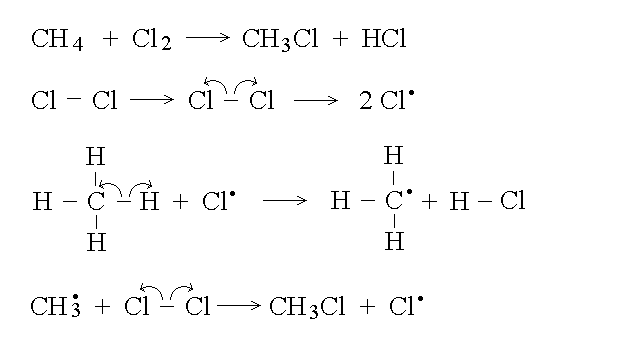
L'addizione al doppio legame avviene in genere con meccanismo elettrofilo: un atomo, ione o molecola con grande tendenza ad attrarre gli elettroni (un "elettrofilo"), cattura un elettrone dell'orbitale di legame π, che viene rotto. Lo stesso atomo usa questo elettrone per combinarsi con uno dei due carboni del doppio legame, mentre l'altro rimane temporaneamente privato di un elettrone (intermedio carbocatione). Il carbocatione reagisce con una specie chimica (in genere uno ione) ricco di elettroni (il "nucleofilo") per ripristinare la configurazione elettronica esterna stabile del carbonio. Un esempio di reazione di questo tipo e' l'addizione di HCl agli alcheni, per la quale si deve considerare che l'HCl e' dissociato in H+ (o H3O+, l'elettrofilo) e Cl- (il nucleofilo):
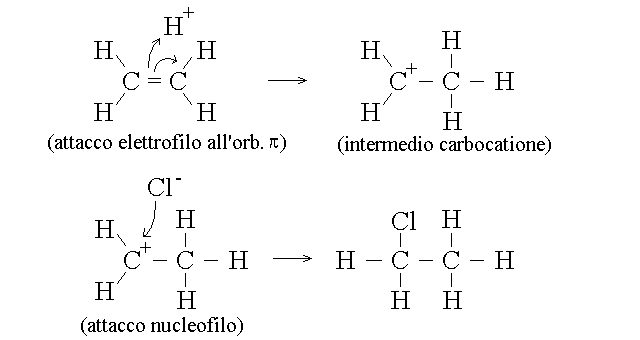
Questo meccanismo di reazione e' possibile perche' l'orbitale di legame π ha una energia relativamente piccola ed e' il punto debole della molecola: l'orbitale di legame di tipo σ non cede i suoi elettroni e pertanto e' resistente all'attacco elettrofilo. e richiede il molto piu' forte attacco radicalico.
L'isomeria, incontrata tanto negli idrocarburi quanto nei composti ternaru del carbonio e' il fenomeno per cui molecole diverse condividono la stessa formula bruta, mentre differiscono nella struttura. L'isomeria e' possibile, ma rara, anche nei composti inorganici (ad esempio hanno la stessa formula bruta gli ioni nitrato e perossinitrito).
Esistono sei tipi di isomeria raccolti in due classi:
a: ISOMERIE DI COSTITUZIONE (STRUTTURALI): (1a) di struttura; (2a) di posizione; (3a) di funzione.
b: ISOMERIE STERICHE: (1b) di conformazione; (2b) geometrica; (3b) ottica.
Di ogni classe occorre conoscere la definizione ed un esempio (cioe' le formule di struttura di due composti che siano tra loro isomeri del tipo considerato).
ISOMERIA DI STRUTTURA (O COSTITUZIONALE). E' quella che deriva dalla eventuale presenza di ramificazioni nella catena di atomi di carbonio che costituiscono la molecola organica; l'esempio piu' semplice e' dato dalle coppie butano-isobutano o butene-isobutene gia' viste in precedenza. Come e' evidente dalle formule di struttura, questi isomeri non possono interconvertirsi tra loro.
ISOMERIA DI POSIZIONE. E' dovuta al fatto che lo stesso gruppo funzionale occupa una posizione diversa nei due isomeri; questo accade ad esempio nella coppia 1-butene-2-butene, o nei tre isomeri dello xilene. E' ancora il caso degli alcoli primari e secondari (ad es. per la coppia 1-propanolo-2-propanolo) o degli idrocarburi nei quali la stessa ramificazione avviene in punti diversi della molecola (ad es. 2-meti pentano e 3-metil pentano):
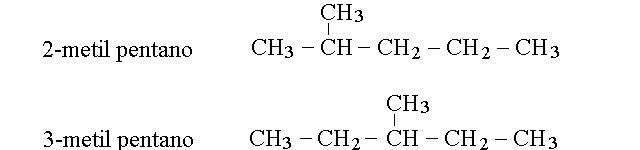
Come e' evidente dalle formule di struttura, gli isomeri di posizione non possono interconvertirsi tra loro.
ISOMERIA DI FUNZIONE. E' in fondo il caso piu' sorprendente perche' le due molecole presentano gruppi funzionali diversi e non condividono nulla tranne la formula bruta. Ad esempio gli alcheni sono isomeri funzionali dei cicloalcani, gli alchini dei cicloalcheni, le aldeidi dei chetoni, gli alcoli degli eteri e gli acidi degli esteri. In genere gli isomeri di funzione non possono interconvertirsi tra loro, con l'eccezione dei tautomeri cheto-enolici.

Un caso particolare di isomeria di funzione, importante nella biochimica, e' la tautomeria cheto-enolica, nella quale i due isomeri (un chetone ed un enolo, vale a dire l'alco di un alchene o di un suo derivato) possono interconvertirsi tra loro, scambiando la posizione di un atomo di idrogeno e di un doppio legame. I due esempi piu' importanti nella biochimica sono quelli dell'acido piruvico e delle basi azotate del DNA e dell'RNA.
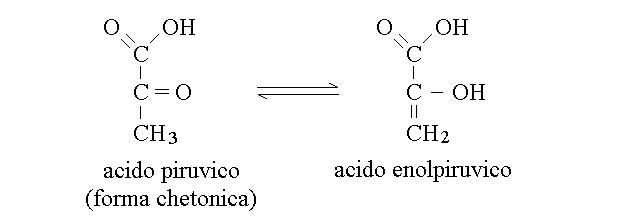
DIFFERENZA TRA LA TAUTOMERIA CHETO-ENOLICA E LA RISONANZA. In alcuni casi possiamo avere una incertezza se una struttura debbe essere considerata un ibrido di risonanza o un tautomero; ad esempio si parla spesso (impropriamente) di tautomeria cheto-enolica delle amidi ed e' effettivamente possibile scrivere una formula dell'amide che presenti il completo trasferimento di uno ione idrogeno dal gruppo =NH2+ all' -O- (si faccia riferimento alla figura degli ibridi di risonanza del gruppo amidico). La differenza tra l'ibrido di risonanza e il tautomero e' questa: nell'ibrido di risonanza si ha riarrangiamento di elettroni e orbitali π (delocalizzazione) ma i nuclei atomici non si muovono e occupano posizioni intermedie tra quelle teoricamente attribuibili ai due o piu' ibridi di risonanza. Nella tautomeria, per contro si ha riarrangiamento non soltanto degli elettroni e degli orbitali π ma anche dei nuclei atomici (in genere un atomo di idrogeno deve spostarsi), e i due tautomeri sono due molecole diverse in equilibrio tra loro, anziche' un'unica molecola con struttura intermedia.
ISOMERIA DI CONFORMAZIONE. Le isomerie steriche sono caratteristiche di composti che sono tra loro molto simili e differiscono per la posizione nello spazio degli stessi gruppi funzionali. L'isomeria di conformazione e' caratteristica del carbonio tetraedrico e ne sono esempi la configurazione sfalsata ed eclissata dell'etano o le configurazioni a sedia e a barca del cicloesano.
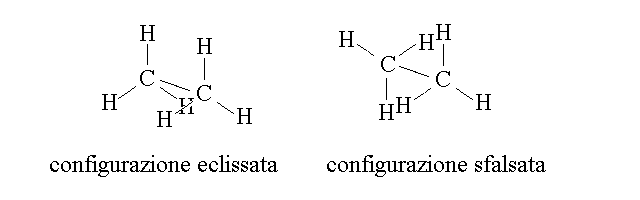
Gli isomeri di conformazione si possono interconvertire piu' o meno facilmente tra loro: infatti negli idrocarburi lineari la rotazione attorno ai legami semplici e' libera (e quindi i conformeri dell'etano sono costantemente in equilibrio tra loro) a meno di impedimenti sterici o di altro tipo (e quindi i conformeri del cicloesano scambiano tra loro piuttosto lentamente).
ISOMERIA GEOMETRICA (o cis-trans). Equivale alla precedente ma e' caratteristica dell'ibridazione sp2, e non ammette la facile interconversione degli isomeri; infatti si deve ricordare che attorno al doppio legame non e' ammessa la rotazione. Poiche' il doppio legame comporta geometria planare, si puo' tirare una riga che passi per questo legame e dividere il piano in due semipiani: nell'isomero cis i due sostituenti si trovano nello stesso semipiano, nell'isomero trans nei due semipiani opposti. Un esempio di questo tipo di isomeria e' stato incontrato nel caso del 2-butene. Ovviamente e' necessario che entrambi i carboni del doppio legame siano sostituiti (cioe' non esiste isomeria geometrica nell'etene o nell'1-butene).
ISOMERIA OTTICA. Si verifica quando ad un atomo di carbonio che presenta ibridazione sp3 (e quindi geometria tetraedrica) sono legati quattro atomi o gruppi differenti. Un atomo di carbonio cosi' configurato e' definito asimmetrico (ed e' indicato in genere con un asterisco quando se ne vuole porre in risalto questa caratteristica). L'esempio piu' semplice e' dato dal 2-butanolo:
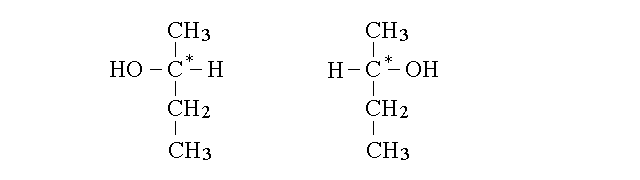
Il carbonio segnato con l'asterisco (carbonio 2) e' asimmetrico e se si facesse ruoatre la molecola di destra il C* non sarebbe sovrapponibile al suo omologo di sinistra (occorre ricordare che la configurazione di entrambi e tetraedrica, non planare, e se si immagina che il c* si trovi nel piano dello schermo due degli atomi che gli sono legati puntano verso l'indietro e due verso l'avanti).
IL CARBONIO ASIMMETRICO. Un atomo di carbonio (o di altri elementi con valenza simile) legato con geometria tetraedrica a quattro atomi o gruppi diversi e' definito asimmetrico o chirale perche' non e' possibile ruotare o riflettere la molecola in modo che appaia uguale a se stessa. Ad esempio, come mostrato in figura, nell'operazione di riflessione, se noi usiamo come piano di riflessione quello individuato dalla posizione dei tre nuclei C, X e Y, osserviamo che l'atomo W si riflette nell'atomo Z; quindi la riflessione non genera una molecola uguale a quella originale. Il carbonio di questo esempio (e di qualunque tipo di isomeria ottica) presenta la stessa asimmetria delle nostre mani (in greco cheir, da cui l'aggettivo "chirale").
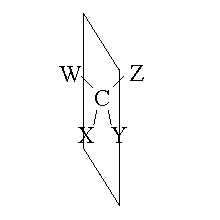
POTERE ROTATORIO DEGLI STEREOISOMERI CHIRALI.
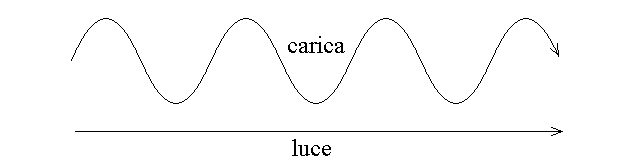
Il carbonio (o qualunque altro atomo) asimmetrico ha la proprieta' di ruotare il piano di polarizzazione della luce polarizzata. Un raggio di luce e' un campo elettromagnetico pulsante che si propaga in linea retta; una carica elettrica (ad es. un elettrone) che si muovesse parallelamente ad esso ne sarebbe alternativamente attratta e respinta e la sua traiettoria sarebbe una sinusoide:
Se noi potessimo osservare il movimento di un certo numero di particlelle cariche poste intorno al fascio di luce che avanza verso di noi, le vedremmo oscillare in tutte le direzioni dello spazio ortogonali alla direzione di propagazione della luce (a sinistra in figura; il fascio di luce che avanza verso di noi e' rappresentato come un punto nero). Questa situazione viene descritta dicendo che i vettori campo elettrico e magnetico oscillano in tutti i piani individuati dalla direzione di propaazione del raggio di luce. Per contro in un raggio di luce polarizzata i vettori campo elettrico e magnetico oscillano in un solo piano e le particelle disposte intorno al raggio si muoverebbero come indicato nell'immagine a destra in figura:
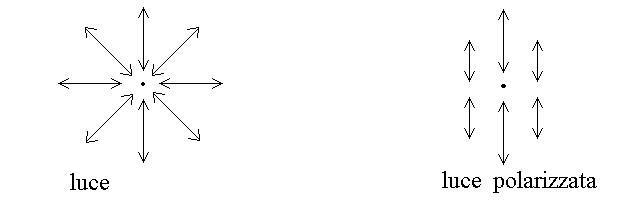
LUCE BIANCA E LUCE MONOCROMATICA. La luce bianca e' una miscela di radiazioni elettromagnetiche di diversa lunghezza d'onda e frequanza (si ricordi la relazione tra questi due parametri: c = λ x ν); la luce monocromatica e' composta invece di radiazioni di una sola lunghezza d'onda e frequenza. Ne' la luce monocromatica ne' quella bianca sono naturalmente polarizzate, ma entrambe possono esserlo se vengono fatte passare attraverso un filtro polarizzatore (il piu' comune filtro polarizzatore e' la lente degli occhiali Polaroid; si trovano inoltre in commercio a basso costo filtri polarizzatori per uso fotografico).
POLARIZZATORI E SPETTROPOLARIMETRI. Se si fa passare un raggio di luce (monocromatica o bianca) attraverso un filtro polarizzatore, la luce che attraversa il filtro e' polarizzata; se la si fa passare attraverso un secondo filtro polarizzatore (filtro analizzatore) allineato con il primo, la luce polarizzata puo' attraversarlo. Se invece il secondo polarizzatore e' ruotato di 90 gradi rispetto al primo, la luce polarizzata non puo' attraversarlo e il raggio viene arrestato. L'esperimento e' schematizzato nella figura seguente:
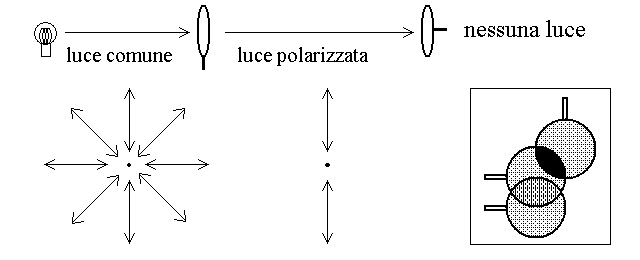
Sulla base dello schema in figura e' possibile descrivere la costruzione dello spettropolarimetro, uno strumento utilizzato per determinare la direzione del piano di polarizzazione della luce polarizzata. In breve, lo strumento consta di una lampada che emette luce bianca; un monocromatore che seleziona una lunghezza d'onda scelta dall'operatore e rende quindi la luce monocromatica; un filtro polarizzatore che rende polarizzata questa luce monocromatica; una cella portacampioni nella quale si mette la soluzione della sostanza da studiare; un filtro analizzatore su supporto ruotabile; ed infine un fotomoltiplicatore che misura l'intensita' della luce che ha attraversato l'analizzatore. In assenza di un campione (o in presenza di un campione contenente sostanze non chirali) la massima intensita' di luce si ottiene quando l'analizzatore e' esattamente allineato col polarizzatore.
Quando il campione posto nello spettropolarimetro e' una soluzione di un soluto organico che presenta il fenomeno dell'isomeria ottica, il piano di polarizzazione della luce polarizzata viene ruotato di un certo numero di gradi verso destra o verso sinistra (potere rotatorio):

A questa proprieta' degli isomeri chirali, e' dovuto un sinonimo della chiralita', quello di isomeria ottica. Vale inoltre la regola che se un isomero chirale ruota il piano di polarizzazione della luce polarizzata verso destra, l'altro isomero lo ruota in direzione opposta della stessa misura e la miscela dei due isomeri in uguale concentrazione e' otticamente inattiva (cioe' non ruota affatto il piano di polarizzazione della luce polarizzata). Una miscela cosi' costituita si chiama racemica.
PROPRIETA' CHIMICHE DEGLI ISOMERI OTTICI. Gli isomeri ottici della stessa sostanza non differiscono rispetto a nessuna proprieta' chimica e fisica "ordinaria": hanno lo stesso punto di fusione, lo stesso pKa se sono acidi, etc. Differiscono naturalmente per il loro potere rotatorio, che e' opposto, ed eventualmente per la loro solubilita' in solventi che siano a loro volta chirali (il 2-butanole e' un esempio di solvente chirale). Per questa ragione in genere la sintesi di laboratorio delle molecole che presentano atomi di carbonio asimmetrici porta alla produzione di miscele racemiche dei due stereoisomeri possibili. Non cosi' la sintesi biologica: nel nostro organismo le molecole vengono sintetizzate da catalizzatori chirali (gli enzimi) e, come accade per i solventi chirali, i catalizzatori chirali sintetizzano in genere uno solo dei due possibili isomeri ottici.
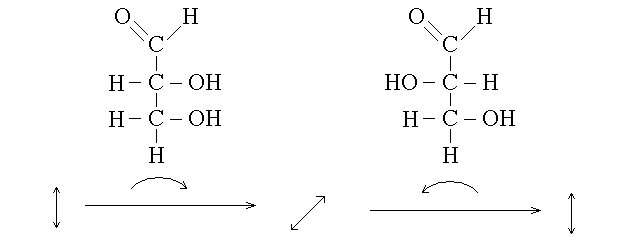
CONFIGURAZIONE D ED L SECONDO FISHER. Fisher, studiando la gliceraldeide (una molecola che contiene tre atomi di carbonio, uno dei quali asimmetrico; nome IUPAC 2,3 di-idrossi propanale), ritenne che fosse possibile assegnare il potere rotatorio destrogiro o levogiro ad una specifica configurazione della molecola, che chiamo' D o L. Studi successivi dimostrarono che la configurazione D di Fisher non corrisponde necessariamente all'isomero destrogiro, ne' la L a quello levogiro. Di conseguenza si usano le lettere D ed L per indicare la configurazione stereochimica del carbonio chirale secondo la convenzione di Fisher e d ed L (o + e -) per indicare il potere rotatorio destrogiro o levogiro.
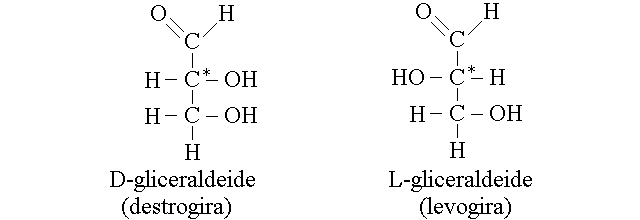
Per rappresentare un atomo di carbonio secondo la convenzione di Fisher occorre procedere come segue: si scrive la molecola in modo che la catena carboniosa principale sia orientata in verticale, con l'estremita' piu' ossidata in alto. Si immagina che il carbonio chirale di interesse sia posto nel piano del foglio di carta e che i carboni che gli stanno sopra e sotto siano profondi rispetto al piano della carta. A questo punto gli altri due sostituenti escono dal piano della carta e vengono verso l'osservatore. Se il sostituente secondario si trova a destra dell'osservatore la molecola ha la configurazione D, altrimenti ha la configurazione L. Per chiarire questa descrizione si possono rappresentare i legami chimici come trangolini che diano l'impressione di profondita' ed indichino quali atomi si allontanano dall'osservatore e quali invece si avvicinino; ad esmepio i due isomeri della gliceraldeide potrebbero essere rappresentati in questo modo:
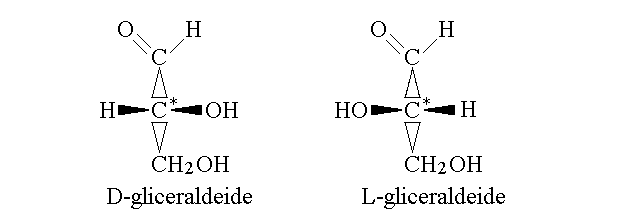
Fisher studiava gli zuccheri, per i quali le regole della sua convenzione erano perfettamente adeguate. Molti composti biochimici anche diversi dagli zuccheri si prestano ad essere descritti in modo soddisfacente utilizzando questa convenzione, che e' pertanto largamente impiegata nella biologia e nella medicina. Esistono pero' composti per i quali la convenzione di Fisher non e' applicabile; ad esempio la catena carboniosa del 2-butanolo non ha una estremita' piu' ossidata, ed e' possibile immaginare composti nei quali non e' ovviamente identificabile un sostituente secondario come l'OH della gliceraldeide. Per questi composti si ricorre ad una convenzione piu' elaborata, che non e' parte del programma di questo corso.
MOLECOLE CON PIU' CENTRI CHIRALI. Molte molecole organiche (e biologiche) presentano piu' di un solo carbonio asimmetrico, e pertanto piu' di due soli stereoisomeri ottici. Come regola generale, e con l'eccezione delle forme meso trattate piu' avanti il numero degli stereoisomeri ttici e' pari a 2n (dove n inidca il numero dei carboni asimmetrici). Ad esempio il glucosio che ha 4 carboni asimmetrici (segnati con un asterisco nella figura) e' uno di 24 = 16 possibili stereoisomeri ottici della stessa formula. Per ragioni pratiche e storiche ciascuno degli isomeri del glucosio ha un suo nome non IUPAC (e questo e' vero per tutti gli zuccheri).
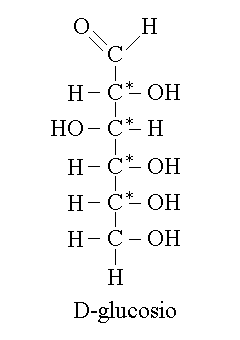
FORME MESO. Un caso interessante si verifica quando una molecola possiede due centri chirali (due carboni asimmetrici) la cui configurazione e' opposta, ed un asse di simmetria centrale, non necessariamente passante per un atomo. In questo caso la molecola si chiama una forma meso (o racemo interno) e non presenta isomeria ottica ne' e' capace di ruotare il piano di polarizzazione della luce polarizzata (cioe' e' priva di attivita' ottica). In una molecola di questo tipo il numero totale di stereoisomeri ottici e' inferiore a 2n. Un esempio di molecola di questo tipo e' l'acido tartarico che presenta due carboni asimmetrici ma ha soltanto tre atomi di stereoisomeri ottici anziche' 4 (=22). Questo accade perche' la molecola presenta un asse di simmetria tale per cui non e' possibile distinguere in essa in modo anequivoco il primo atomo di carbonio dall'ultimo; di conseguenza risultano distinte le configurazioni 2D,3D, 2L,3l e 2D,3L che e' uguale alla 2L,3D. La molecola non puo' essere rappresentata in modo completamente anequivoco seguendo la convenzione di Fisher, pertanto nella figura aggiungo accanto ad ogni carbonio chirale la definizione della sua configurazione. Come e' evidente dalla figura, ruotando su se stessa la configurazione 2D,3L si ottiene la sua inversa 2L,3D. Questo accade perche' in realta non e' possibile dire in modo anequivoco quale sia il C1 e quale sia il C4; infatti la moleocla ha un piano di simmetria che passa attraverso il legame tra il C2 ed il C3 (qualunque numerazione si scelga).
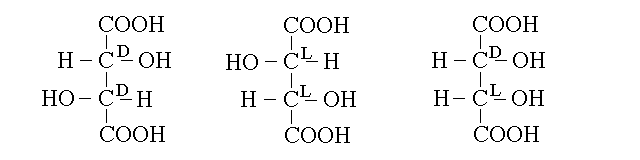
Per comprendere meglio questo fenomeno, si puo' confrontare l'acido tartarico (2,3 diidrossi butanodioico) con l'acido 2,3 diidrossi butanoico, che ha due centri chirali e 22=4 stereoisomeri. Infatti e' evidente che nella seconda molecola, al contrario della prima l'identificazione del C1 e' anequivoca e non ci sono assi di simmetria intramolecolari; conseguentemente gli isomeri 2D,3L e 2L,3D sono due molecole perfettamente distinte:
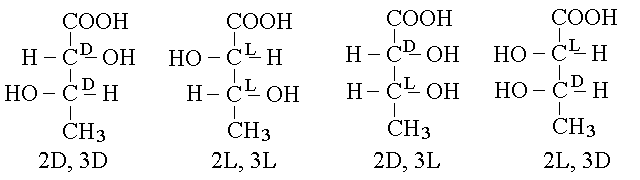
Torna a: didattica; Medicine non Scientifiche; Biblioteca Digitale.